
Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR), which is considered to be an immune system for bacteria, has been widely used as a tool for genome editing and genotyping. It has also been reported to be associated with virulence factors in some bacteria. To understand the role of CRISPR in the virulence and evolution of pathogenic Vibrio parahaemolyticus, 154 V. parahaemolyticus strains isolated from clinical samples and 54 strains from food samples taken in Shenzhen, China were subjected to a correlation analysis of CRISPR and virulence factors TDH and TRH. We also performed multilocus sequence typing (MLST) for genotype analysis. Six different CRISPR sequence types (CSTs) of V. parahaemolyticus were identified, and CSTs were found to be significantly associated with the virulence factors tested and MLST genotype. Therefore, CSTs provide insight into the evolution of V. parahaemolyticus. Moreover, identification of CSTs may lend insight into the virulence potential of strains.
Introduction
V
Molecular typing has been increasingly used to characterize isolates associated with outbreaks. Several molecular typing methods, including ribotyping, pulsed-field gel electrophoresis, and multilocus sequence typing (MLST), have been used to analyze bacterial homology and their molecular evolution (Maiden et al., 1998; Bag et al., 1999; Marshall et al., 1999). In 2008, Gonzalez-Escalona et al. developed the standard MLST scheme for V. parahaemolyticus (
Clustered regularly interspaced short palindromic repeats (CRISPR) is known as an immune system for bacteria. Together with cas (CRISPR-associated) genes, CRISPR specifically cleaves invasive exogenous plasmids and bacteriophage double-stranded DNA (Garneau et al., 2010). The interaction between the CRISPR/Cas system of V. cholerae and bacteriophage has been previously described (Seed et al., 2013). In addition to immune functions, it has been reported that the CRISPR/Cas system might also play an alternative role in the regulation of virulence and genome evolution in pathogenic bacteria (Westra et al., 2014). Moreover, CRISPR has also been widely used for subtyping Salmonella in order to improve laboratory surveillance (Fabre et al., 2012; Shariat et al., 2013). However, to date only one possible CRISPR/Cas sequence for V. parahaemolyticus has been listed in the CRISPR database. Based on BLAST results from the CRISPR database (
Materials and Methods
Bacterial strains and DNA extraction
A total of 208 representative isolates of V. parahaemolyticus were obtained during 2006–2011 in Shenzhen, China. One hundred fifty four clinical isolates collected from laboratory-based sentinel surveillance of acute diarrheal disease and V. parahaemolyticus foodborne disease outbreaks in Shenzhen had various 25 serotypes, whereas 54 food isolates recovered from oysters, fish, shellfish, red-stewed duck, fried pork, and beef possessed different 35 serotypes in which 14 serotypes were the same as the clinical isolates (Table 1 and Supplementary Table S1; Supplementary Data are available online at
52 different MLST sequence types included.
37 different serotypes included.
ST262, O1:K69 (n=4, three are clinical isolates and one is a food isolate).
CCs, clonal complexes; CRISPR, clustered regularly interspaced short palindromic repeats; MLST, multilocus sequence typing.
Detection of virulence factors
Real-time polymerase chain reaction (PCR) was performed on each isolate to detect the virulence factors, including tdh, trh, and the V. parahaemolyticus marker gene tlh on an ABI 7500 real-time PCR system (Applied Biosystems, Inc., Foster City, CA). Primers and the Taqman probe for tdh were modified slightly from the sequences described by Blackstone (Blackstone et al., 2003). The forward primer (5′-AAACATTTGCCTTTGAGCTTCCA-3′), reverse primer (5′-CTCGAACAACAAACAATATCTCATCAG-3′), and TaqMan probe (5′-FAM-CCGGGGTGTCCCTTTTCCTGCCCCCGG-DABCYL-3′) were used to detect tdh gene. The trh and tlh genes were identified as previously described by Davis (Davis et al., 2004) and Ward (Ward and Bej, 2006), respectively.
MLST
V. parahaemolyticus MLST analysis is described on the PubMLST website (
PCR amplification and genotype for CRISPR
The sequence of CRISPR from V. parahaemolyticus was obtained from the CRISPR website (
Statistical analysis
The association of CRISPR with virulence factor tdh and CCs was analyzed using a 2×2 table and SPSS 15.0 software.
Results
Distribution of virulence factors
We found that 94.81% of clinical isolates (146/154) contained tdh, whereas 12.96% (7/54) of the food isolates were tdh positive. The 202 isolates were trh negative. Six clinical isolates, including O1:K69 (n=4), O4:K12, and O2:K28 serotypes, were positive for both the trh and tdh genes (Table 1).
Analysis of STs and CCs
A total of 79 unique MLST STs were identified among the 208 V. parahaemolyticus isolates, among which 55 STs were novel in the PubMLST database (Supplementary Table S1). ST-3 was the most common ST, which contained 84 isolates (40.4%) and spanned multiple serotypes. Eight other STs were represented by three or more isolates: ST-265 (n=15), ST-120 (n=12), ST-8 (n=9), ST-262 (n=4), ST-68 (n=3), ST-216 (n=3), and ST-332 (n=3). Forty-eight STs and 41 STs were identified in 54 food isolates and 154 clinical isolates, respectively, which exhibited greater diversity in food isolates. In our study, compared to the PubMLST database, 6 clonal complexes (CCs) and 10 potential clusters were identified (Supplementary Table S1 and Supplementary Fig. S1). Six CCs included CC3 (n=91), CC8 (n=10), CC120 (n=14), CC345 (n=17), CC527 (n=2), and CC36 (n=1) (Supplementary Figure S1 and Supplementary Table S1).
All isolates from the six CCs and their SLVs were tdh positive (Supplementary Table S1). Among CC3, the largest serotype was O3:K6, which accounted for 67 strains (73.6%). Of these, 64 were included in ST-3 and the remaining 3 originated from SLVs of ST-3. CC8 included the founder ST8 and its SLV was identified as serotype O1:K56. The serotype of all isolates of CC120 was O3:K29 and all isolates in CC345 were identified as serotype O4:K8. In addition, ST-527 and ST-484 in CC527 were identified as serotype O3:K41 and O1:K41, respectively.
Analysis of CSTs
Six CRISPR sequence type (CST) patterns were identified, including CST0 (n=59), CST1 (n=3), CST2 (n=15), CST3 (n=91), CST4 (n=27), and CST5 (n=13) (Table 1 and Fig. 1). Isolates that lacked CRISPR were grouped into CST0. CST1 and CST2 contained only one spacer, while CST3 and CST4 contained two with a single nucleotide polymorphism (SNP) (A/G) present in the first spacer. The sequence of CST3 was identical to that in the reference strain of V. parahaemolyticus RIMD 2210633. Compared to CST4, one more spacer was identified in CST5.
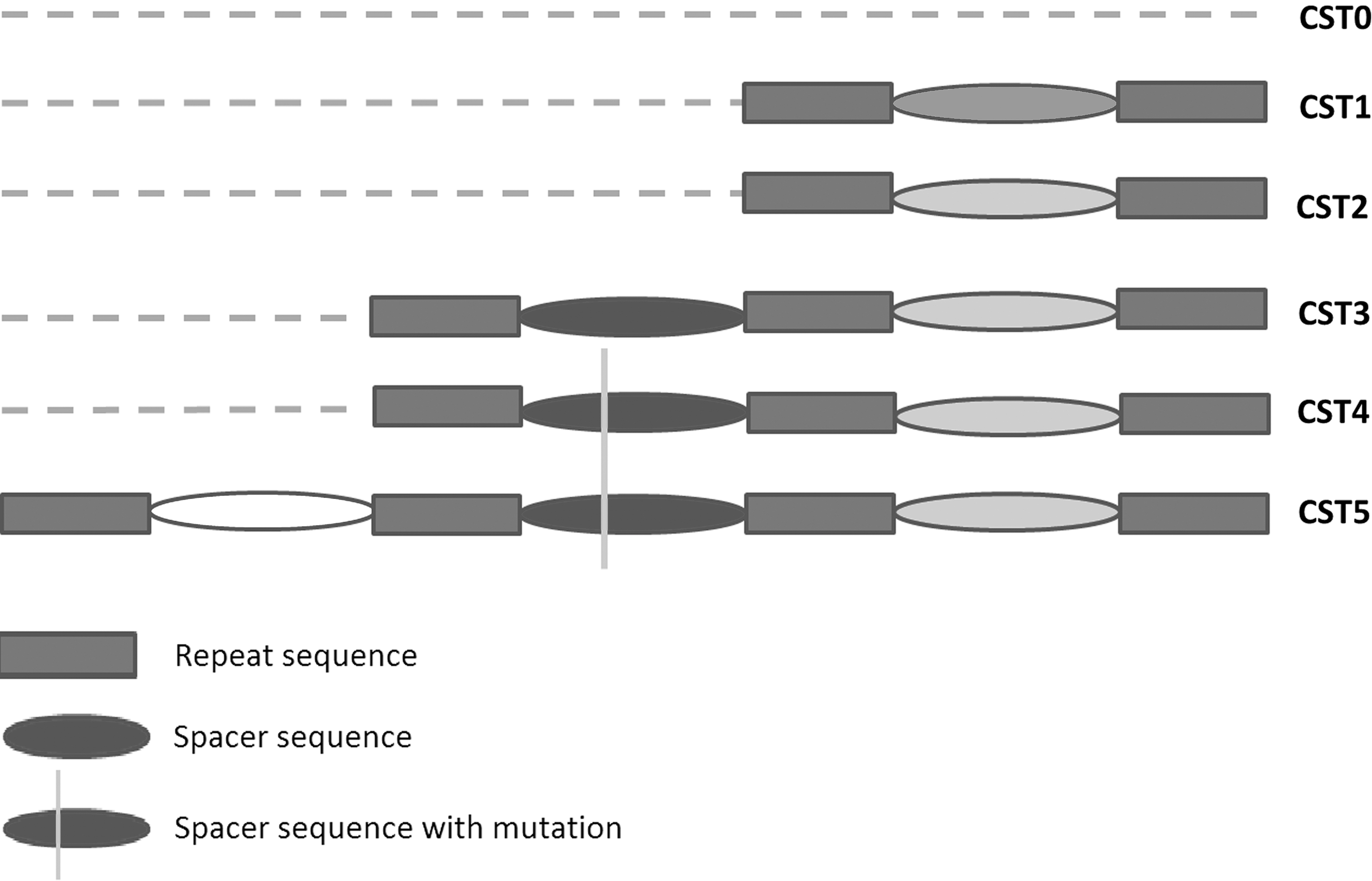
Six different clustered regularly interspaced short palindromic repeats sequence type patterns identified in 208 Vibrio parahaemolyticus isolates from clinical and food-associated samples.
Correlation between CSTs and virulence factors
Among 208 isolates, there were 153 tdh-positive isolates and 55 tdh-negative isolates. Among 153 tdh-positive isolates, 149 isolates were identified as CRISPR positive, while 55 tdh negative isolates were identified as CRISPR negative (CST0). The κ correlation was 0.952, which had significant association between CRISPR and virulence factor tdh (Table 2). Four tdh positive isolates that were identified as CRISPR negative were O1:K69 serotype, trh positive and ST-262.
CRISPR, clustered regularly interspaced short palindromic repeats.
Correlation between CSTs and MLST genotype
CST3, which only differed from CST4 by a single SNP, was significantly associated with clonal complex 3 (CC3) (Table 1). All isolates from CST3 were grouped into ST3 (n=83) and the single-locus/double-locus variants (SLV/DLV) of ST3 (n=7). Compared to CST3, CST4 was more diverse with regard to the genotype. In CST2, the sequence types were mainly composed of ST8 (O1:K56, n=8) and ST120 (O3:K29, n=4). CST5 had three spacers and covered ST120 (O3:K29, n=8) and ST332 (O4:K9, n=3). Although only six CRISPR sequence type patterns were observed in V. parahaemolyticus in our study, a significant association between the CSTs and CCs was found and the κ correlation was 0.845 (Table 2). In particular, the CST3 was completely concordant with CC3. Compared to CST4, only a single difference in nucleotide sequence (A/G) occurred in one spacer in CST3, which provides a novel and efficient marker for those pandemic pathogens in CC3.
Discussion
CRISPR is mainly regarded as an adaptive immune system in prokaryotes; however, recent studies have identified involvement in other important processes, including the regulation of virulence genes, biofilm formation, and genome editing and genotyping (Cady and O'Toole, 2011; Sampson et al., 2013; Vercoe et al., 2013). Although there is no direct evidence of how CRISPR affects tdh, the significant association between virulence factor tdh and CRISPR/Cas shown in this study suggests a close relationship between these components in V. parahaemolyticus. In our study, four tdh positive isolates were negative for the CRISPR sequence, which may be due to a survival advantage in the intestinal environment through evolution into diverse CST types through procurement of an exogenous spacer sequence. Furthermore, the four isolates (ST262, O1:K69 serotype) may represent emerging pathogenic V. parahaemolyticus strains that have not as yet developed into other CST types. Type II CRISPR/Cas systems have been shown to play a role in virulence (Sampson et al., 2013), and therefore future studies are needed to address the involvement of subtype I-F CRISPR/Cas with csy3 in the virulence of V. parahaemolyticus.
MLST has been used to analyze evolution of bacterial species. In order to analyze the association of CRISPR/Cas with MLST, the 208 isolates were typed by MLST. It is worth noting that CC120 and CC527 were identified when the newly discovered SLV ST-480 and ST484 isolates were added to the PubMLST database, respectively. All 6 CCs had a CST, and furthermore all 91 of the isolates in CC3 possessed CST3, indicating that the occurrence of CST3 was completely consistent with CC3. So CST3 can be used as an MLST marker for molecular typing instead of CC3. Our results also showed that all CCs that we identified from the PubMLST database were present in clinical and food isolates possessing the virulence-related tdh gene, while none of the clonal complexes were found in the strains lacking the tdh gene. These results suggest that the isolates that were tdh positive were responsible for the development of the epidemic population. The concordance between virulence and CCs could also be caused by lateral acquisition of genes for virulence determinants (Yan et al., 2011). In addition, all strains that clustered into the same ST possessed the tdh gene, which seems to contradict previous results showing that virulence genes are not related to STs (Chao et al., 2011; Yu et al., 2011).
An increase in the type and number of spacers within the CRISPR/Cas system occurs when the bacteria incorporate short sequences from invading bacteriophages or plasmids into the CRISPR sequence (Karginov and Hannon, 2010). Therefore, to some extent, the number of spacers in CRISPR might reflect the degree of evolution of the species. Considering the small number of spacers present, V. parahaemolyticus is believed to be in the initial stages of evolution. All food-associated isolates lacking tdh were CST0, which suggests that environmental strains might be in the early stages of evolution. The clinical isolates contained more spacers, implying that this adaptation evolved with a clear fitness advantage in the host. CST5 was the most evolved group and was mainly composed of ST120 (O3:K29) and ST332 (O4:K9), which were recently reported as modern epidemic serotypes in China (Qu et al., 2012). CST4 only had a single SNP compared to CST3 and one spacer less compared to CST5. In addition, several potential semiclonal complexes were present in CST4. Therefore, we hypothesize that CST3 and CST5 may have directly evolved from the CST4 group. The CC3, which contained O3:K6 and its serovariants, has been detected throughout the world as a pandemic pathogen (Nair et al., 2007; Chao et al., 2011; Velazquez-Roman et al., 2013). Similarly, the CST3 group has become the pandemic V. parahaemolyticus strains in China; it remains to be determined whether the CST5 group will develop a pandemic strain.
In conclusion, in this study we analyzed the diversity of CRISPR in V. parahaemolyticus together with virulence factors and MLST genotypes. The association of CRISPR between tdh gene and genotype will provide new approaches for monitoring pathogenic V. parahaemolyticus in clinical and foods samples. Moreover, the CRISPR sequence type could be used as a potential marker for identification of virulence factor tdh and CC3 in V. parahaemolyticus.
Footnotes
Acknowledgments
This research was supported by the China National Science and Technology Major Projects Foundation (No. 2012ZX10004215-003-005 to Q.H.) and Natural Science Foundation of China (No. 81071433 to Q.H.).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.