
Abstract
The distribution of 18 staphylococcal enterotoxin (SE) or SE-like (SEl) genes in Staphylococcus aureus strains from different sources in east China was investigated. Among all 496 S. aureus strains, 291 strains carried one or more SE genes. The more frequently occurred genes were sea, seb, seg, selk, sell, selm, selo, and seq; the less frequent occurred genes were sec, selj, and ser. The classic SE genes and the enterotoxin gene cluster (egc) (seg, sei, selm, seln, selo, and/or selu) accounted for 25.67% and 61.68% of all detected genes, respectively. There were three gene clusters (egc, sea-sek-seq, and sed-sej-ser), of which the egc cluster was the important one that could generate novel complexes, and the sea-sek-seq cluster was a close relative to the hospital-acquired methicillin-resistant S. aureus. The SE gene distributions were different among strains of different sources and formed diverse toxin gene profiles. The human- and foodborne-origin strains harbored classic and novel SE and SEl genes, whereas animal-origin strains harbored egc and other novel SE and SEl genes mainly. The foodborne- and human-origin strains were the main dangerous factors of classic staphylococcal foodborne poisoning, whereas the strains (especially from animals) that carried egc and other novel genes mainly should be new potential dangerous factors for food safety.
Introduction
E
Previous studies suggested that most of SFP outbreaks (SFPOs) are caused by five classical SEs, whereas the hazards from novel types of SEs and SEls have not been given more recognition (Omoe et al., 2002). This study aims to investigate the distribution of SE and SEl genes of S. aureus strains from different sources in east China.
Materials and Methods
Strains
A total of 496 S. aureus strains were collected in east China during 2007–2014, including 310 strains isolated from humans in local tertiary hospitals, 3 strains isolated from knives and tools, such as hairdressing razors and pedicure razors, 74 strains isolated from food of farmers' markets or supermarkets, 35 strains from raw milk from distinct farms, and 74 strains isolated from animals, including pigs, chickens, ducks, geese, dogs, sheep, and peacock in animal hospitals. All strains were identified by using the hemolysis test on blood agar, the coagulase test, and various biochemical tests with the ATB ID32 STAPH test system for identifying staphylococcus species (BioMerieux, Lyon, France).
Extraction of bacteria genomic DNA
All the S. aureus strains were stored at −70°C and cultivated on Baird-Parker agar. A single colony was inoculated in 4 mL of Lysogeny broth (LB) liquid medium at 37°C overnight, and 1 mL of the culture was extracted with a Takara bacteria genomic DNA extraction kit (Takara, Kyoto, Japan).
Multiplex polymerase chain reaction assay
Eighteen pairs of primers for the SE and SEl genes of S. aureus were used in multiplex polymerase chain reaction (PCR) assays (Table 1). The primers were divided into seven groups based on the different SE and SEl gene lengths. Each assay contained 1 μL of prepared DNA template, 2.5 μL of 10× EasyTaq Buffer (Takara), 2.5 μL of 2.5 mM deoxynucleotide triphosphates (Takara), 0.5 μL of several upstream and downstream primers (10 μM), and 0.25 μL of DNA polymerase (5 U/μL) (Takara), and the final system volume was adjusted to 25 μL. The PCR condition included the same steps as follows: predenaturation at 94°C for 5 min, 30 cycles of denaturation at 94°C for 30 s, annealing at 58°C for 30 s (annealing temperature decreases by 0.5°C each cycle), extension at 72°C for 60 s, and final extension at 72°C for 10 min.
The PCR primer groups were able to amplify the target genes with multiplex PCRs. Nonspecific bands were not observed, and the sizes of all the amplified bands were easy to differentiate (Fig. 1). The sequencing of the extracted PCR product was performed by GenScript (Nanjing, China) and the data were analyzed using the DNAstar software. Amplicon sequences were analyzed with DNAstar software to verify against S. aureus database sequences to ensure specificity.
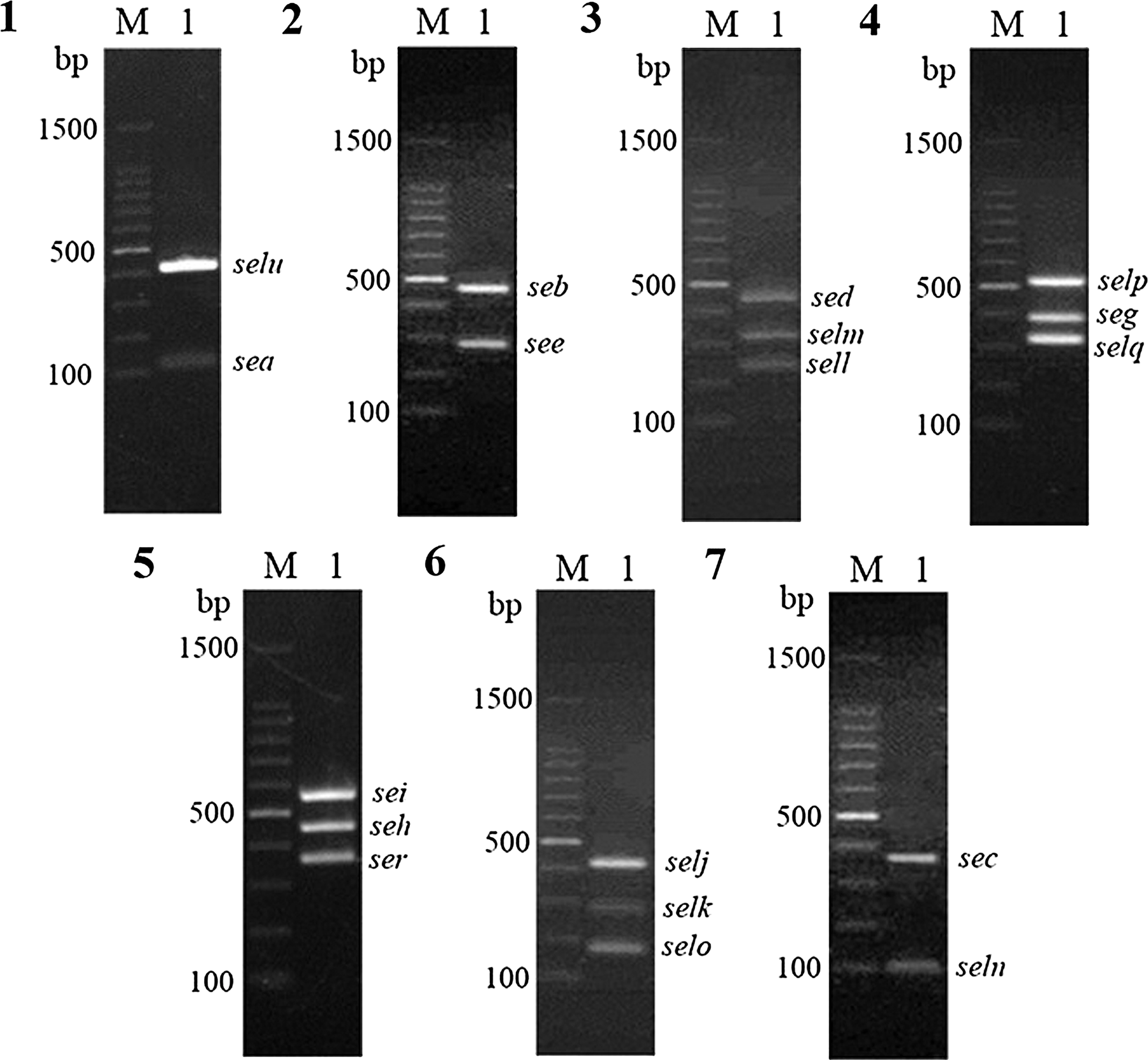
Seven multiplex polymerase chain reaction results of SE and SEl genes. (1) sea (127 bp), selu (410 bp). (2) see (286 bp), seb (477 bp). (3) sell (240 bp), selm (300 bp), sed (451 bp). (4) selq (330 bp), seg (400 bp), selp (537 bp). (5) ser (368 bp), seh (463 bp), sei (576 bp). (6) selo (180 bp), selk (282 bp), selj (426 bp). (7) seln (103 bp), sec (371 bp). SE, staphylococcal enterotoxin; SEls, staphylococcal enterotoxin-like toxins.
Results
Among all 496 S. aureus strains, 291 strains (58.67%) carried one or more SE genes. The genes sea, seb, sec, sed, see, seg, seh, sei, selj, selk, sell, selm, seln, selo, selp, selq, ser, and selu were found in 19.35%, 11.69%, 4.23%, 2.62%, 4.64%, 19.15%, 4.64%, 9.68%, 1.81%, 10.89%, 10.98%, 13.91%, 6.45%, 19.15%, 4.44%, 13.10%, 2.92%, and 7.06% of all strains, respectively. The classic genes and egc (seg, sei, selm, seln, selo, and/or selu) accounted for 25.67% (211/822) and 45.50% (374/822) of all detected genes, respectively. The genes of egc cluster with the coexisted 133 genes accounted for 61.68% (507/822) of all detected genes. Four strains (0.81%) carried completed egc cluster (seg, sei, selm, seln, selo, selu) and 49 strains (9.88%) carried the 4 or 5 genes of the egc cluster simultaneously. Thirty-three strains (6.65%) carried sea, sek, and seq genes simultaneously. Nine strains (1.81%) carried the sed, selj, and ser genes simultaneously.
There were 54.19% (168/310) clinic strains that carried one or more SE genes; the genes sea, seb, sec, sed, see, seg, seh, sei, selj, selk, sell, selm, seln, selo, selp, selq, ser, and selu were found in 21.29%, 10.65%, 5.81%, 1.94%, 2.58%, 17.74%, 1.29%, 10.97%, 1.29%, 13.87%, 10.97%, 11.61%, 6.27%, 15.48%, 4.19%, 16.13%, 1.61%, and 5.81% of 310 strains, respectively; the genes of the classic genes accounted for 26.41% (131/496), whereas the genes of egc cluster accounted for 42.74% (212/496). About 62.16% (46/74) strains from animals (pigs, chickens, ducks, goose, dogs, sheep, and peacocks) harbored one or more genes; the genes seb, sec, sed, selj, seln, selp, and ser were absent; the other genes sea, see, seg, she, sei, selk, sell, selm, selo, selq, and selu were found in 10.81%, 1.35%, 29.73%, 2.70%, 9.46%, 5.41%, 5.41%, 24.32%, 33.78%, 10.81%, and 20.27% of 74 strains, respectively; the genes of classic SE genes and the genes of the egc cluster accounted for 7.76% (9/116) and 75% (87/116), respectively. There were no SE or SEl genes existing in three knives and tools source strains. About 70.64% (77/109) of 109 foodborne strains from raw milk and food harbored one or more SE genes; the genes sea, seb, sec, sed, see, seg, seh, sei, selj, selk, sell, selm, seln, selo, selp, selq, ser, and selu were found in 20.18%, 25.94%, 2.75%, 6.42%, 12.84%,16.51%, 15.60%, 6.42%, 4.59%, 6.42%, 12.84%, 11.93%, 10.09%, 20.18%, 8.26%, 6.42%, 6.42%, and 1.83% of 109 strains, respectively; the genes of classic genes and the genes of the egc cluster accounted for 33.81% (71/210) and 34.76% (73/210), respectively (Table 2).
SE, staphylococcal enterotoxin; SEls, staphylococcal enterotoxin-like toxins.
Discussion
The most notable virulence factors, SEs, of S. aureus could cause the sporadic food-poisoning syndrome or foodborne outbreaks. The prevalence rates were different in different articles, as strains were from geographically diverse locations. Varshney et al. (2009) reported that 99% clinic strains harbored at least one SE gene. Srinivasan et al. (2006) reported that 93.6% strains from milk of cow with mastitis harbored one or more SE genes. In this study, 18 kinds of SE genes were tested and diverse gene patterns were found in S. aureus strains from different sources of human, animals, food, and milk. About 60% strains carried one or more SE genes. About 55% human-origin, 62% animal-origin, and 70% foodborne strains harbored one or more SE genes. These results showed that S. aureus strains from different geographic regions and different sources have different SE gene profiles. The more frequently occurred genes were sea, seb, seg, selk, sell, selm, selo, and seq (all >10%), the less frequently occurred genes were sec, selj, and ser (all <3%). About 46% SE and/or SEl genes belonged to the egc cluster, which often co-occurred with other genes or gene cluster, such as sea, seb, sell, sea-sek-seq.
It is noteworthy that the occurrence of SE genes is both relatively abundant and diverse in strains of three different sources: human, animals, and foods and raw milk. It is interesting to note that the prevalence of genes in foodborne strains was higher than that in human-origin strains (χ2 = 5.63, p < 0.05), but there was no difference between human and animal-origin strains (χ2 = 0.043, p > 0.05), also between animal and foodborne strains (χ2 = 3.53, p > 0.05), which was caused by the distribution difference of classic SE, egc, and the other genes in strains of the three different sources. The number of classic SE genes in foodborne strains was significantly higher than in human-origin strains (χ2 = 16.09, p < 0.01) and significantly higher than in animal-origin strains (χ2 = 31.06, p < 0.01). This suggested that the foodborne- and human-origin strains were the main potential cause of classic SFP. In contrast, the genes of the egc cluster, seg, selm, selo, and the other genes, selq, selk, sell, were found especially in animal-origin and foodborne-origin strains; there was no significant difference with the number of egc genes between human-origin and boodborne-origin strains, but the number of egc genes in animal-origin strains was significantly higher than in human-origin strains (χ2 = 21.32, p < 0.01) and higher than in foodborne strains (χ2 = 15.00, p < 0.01). It seemed that these animals had little contact directly with humans, resulting in the less transmission of toxin genes between them. A previous study has shown that strains that carried novel SE or SEl genes could cause SFPOs (McLauchlin et al., 2000), so these strains (especially from animals) should be a new potential threat to food safety.
There were three gene clusters egc, sea-sek-seq, and sed-sej-ser in this study; these clusters often coexisted with other SE genes or gene clusters. Previous studies have shown that one of the egc genes usually indicates the presence of all egc genes, and the egc cluster could generate novel complexes by reproduction or exchanging with other strains (Jarraud et al., 2001; Thomas et al., 2007). In this study, complete egc clusters occurred only in a few strains, most of the strains contained the incomplete egc clusters lacking one or more genes and coexisted with other genes or gene clusters, such as sea, seb, sec, sek, seq, and so on. (data not shown), which confirmed that the egc locus could be the source of enterotoxin gene evolution (Jarraud et al., 2001; Thomas et al., 2006). Another important cluster was sea-sek-seq, our study showed that the cluster was a close relative to hospital-acquired (HA) methicillin-resistant S. aureus (MRSA)-III clone (unpublished data). The cluster sed-selj-ser was of less occurrence and often co-occurred with the egc cluster, which shows that the cluster was not popular in S. aureus strains in east China.
Conclusion
About 60% strains from different sources in this study harbored one or more SE genes, the more frequently occurring genes were sea, seb, seg, selk, sell, selm, selo, and seq. The SE gene distributions were different among strains of different sources and formed different distribution patterns. There were three gene clusters egc, sea-sek-seq, and sed-sej-ser, of which the egc cluster was the important cluster that could generate novel complexes, and the sea-sek-seq cluster was a close relative to the HA-MRSA-III clone. The human- and foodborne-origin strains harbored classic and novel SE and SEl genes, whereas animal-origin strains harbored mainly egc genes. Human and foodborne strains were the main potential cause of classic SFP, whereas the strains (especially from animals) that carried novel SE and SEl genes could become new potential dangerous factors for food safety.
Footnotes
Acknowledgments
This work was supported by grants from the Fundamental Research Project of Jiangsu High Education Institutions (15KJA230001), the Jiangsu Agriculture Science and Technology Innovation Fund [CX(14)2088], the Natural Science Foundation of Yangzhou (YZ2014060), the China Agriculture Research System (CARS-41-K08), the Priority Academic Program Development of Jiangsu Higher Education Institutions, and the National Science and Technology Support Plan (2012BAK17B10).
Disclosure Statement
No competing financial interests exist.