
Abstract
Methods to rapidly identify serotypes of Salmonella enterica subspecies I are of vital importance for protecting the safety of food. To supplement the serotyping method dkgB-linked intergenic sequence ribotyping (ISR), single-nucleotide polymorphisms were characterized within adenylate cyclase (cyaA). The National Center for Biotechnology Information (NCBI) database had 378 cyaA sequences from S. enterica subspecies I, which included 42 unique DNA sequences and 19 different amino acid sequences. Five representative isolates, namely serotypes Typhimurium, Kentucky, Enteritidis phage type PT4, and two variants of Enteritidis phage type PT13a, were differentiated within a microsphere-based fluidics system in cyaA by allele-specific primer extension. Validation against 25 poultry-related environmental Salmonella isolates representing 11 serotypes yielded a ∼89% success rate at identifying the serotype of the isolate, and a different region could be targeted to achieve 100%. When coupled with ISR, all serotypes were differentiated. Phage lineages of serotype Enteritidis 13a and 4 were identified, and a biofilm-forming strain of PT13a was differentiated from a smooth phenotype within phage type. Comparative ranking of mutation indices to genes such as the tRNA transferases, the diguanylate cyclases, and genes used for multilocus sequence typing indicated that cyaA is an appropriate gene for assessing epidemiological trends of Salmonella because of its relative stability in nucleotide composition.
• Allele‐specific primer extension (ASPE) was validated as a subtyping method for Salmonella enterica by correctly identifying single‐nucleotide polymorphisms (SNPs) in the cyaA gene from 25 strains isolated from the environment of poultry.
• More than 80 SNPs in the cyaA gene of Salmonella were tabulated, which indicates that cyaA is useful as a gene target for ASPE intended to provide some subtype information after the serotype is assigned by intergenic sequence ribotyping.
• Genes such as cyaA that have a nonsynonymous to total (NS/T) mutation index of less than 0.5 may be more conservative for establishing subtype within serotype than genes with a higher NS/T index.
• Genes with higher NS/T indices, such as the diguanylate cyclases, might be more sensitive for detecting rapidly emerging strain heterogeneity.
Introduction
A 42-
Other genome-based methods have been developed to serotype the salmonellae in addition to ISR, and these include pulsed-field gel electrophoresis (Swaminathan et al., 2001; Ji et al., 2006), amplified fragment length polymorphism (Liebana et al., 2002), multilocus sequence typing (MLST) (Sukhnanand et al., 2005; Ji et al., 2006; Liu et al., 2011), multiple-locus variable-number tandem repeats (Lindstedt et al., 2004), CRISPR analysis (Fabre et al., 2012), and repetitive extragenic palindromic polymerase chain reaction (PCR) (Versalovic and Lupski, 2002). However, most of these methods do not correlate specific biological properties with SNPs that emerge between closely related strains within serotypes. For S. enterica, examples of using SNPs to pathotype S. enterica include (1) association of 16 SNPs with transition from an invasive phenotype to one that is environmentally prevalent, but epidemiologically inconsequential (Guard et al., 2011), (2) impact on the predicted epitopes of flagella (McQuiston et al., 2011), and (3) impact on antibiotic resistance (Song et al., 2010). The importance of SNPs is emphasized by a fundamental concept of microevolution; specifically, a single base pair change can have as much impact on the ability of a pathogen to cause disease as does an exchange of chromosomal DNA of thousands of base pairs. An example of such microevolution is in Listeria monocytogenes, where SNPs were used to identify outbreak strains and to determine their incidence in ready-to-eat foods relative to environmental strains (Ducey et al., 2007; Ward et al., 2008; Van Stelten et al., 2010).
One physiologically important gene that is suggested as a target for microevolutionary studies in S. enterica serotypes is adenylate cyclase (cyaA) ((Zhang et al., 1996; Morales et al., 2007). It has been associated with evolutionary trends in avian-adapted serotypes, such as Salmonella serotypes Pullorum and Gallinarum (Morales et al., 2007). Several other factors make cyaA an attractive gene target for subtyping. These factors are as follows: (1) cyaA is central to energy production and metabolism (Lory et al., 2004), (2) required for virulence (Curtiss III and Hassan, 1996), (3) it is present as one copy, and (4) it is associated with physiological change when mutated (i.e., reduced lethality, growth, and environmental persistence) (Kennedy et al., 1996; Zhang et al., 1996). In addition, the mutations within cyaA gene have potentially evolved as a coping mechanism during changing environmental conditions, allowing S. enterica serotypes to maintain critical functions required for baseline survival and infection potential (Aravind and Koonin, 1999; Wolfgang et al., 2003; Baker and Kelly, 2004). To further understand if there is any impact by selecting cyaA over other genes for developing SNP analysis supporting ISR serotyping, BLAST analyses were used to collect pertinent information for characterization of three other sets of genes. Set 1 included 20 tRNA transferases (Ogle and Ramakrishnan, 2005), set 2 included 13 diguanylate cyclases (DGC) (Jenal and Malone, 2006), and set 3 included 23 genes commonly included in other genome studies. Results suggest that cyaA has characteristics favorable for supporting ISR and for being a reference gene for assessing heterogeneity occurring between strains associated with outbreaks.
Materials and Methods
BLAST for recovery of available S. enterica subspecies I sequences
A strategy for BLAST analysis was followed to find only unique cyaA sequences and then to translate those sequences into unique amino acid sequences. The reference genome for all BLAST searches was Salmonella enterica serovar Typhimurium LT2 (NC_003197.1). Specifically, gene STM3939 was used in BLAST searches of complete and draft genomes to obtain all cyaA sequences of record within S. enterica subspecies I between August 15 and September 20, 2015. The National Center for Biotechnology Information (NCBI,
Gene cyaA is highly conserved within the genome of S. enterica, and there is only one copy (McClelland et al., 2001). To determine how the nonsynonymous to total (NS/T) index of other genes compared to cyaA, three sets of genes were similarly analyzed (Fig. 1), and details for each gene are listed in Table 1. The tRNA transferases (Ogle and Ramakrishnan, 2005) and DGCs (Jenal and Malone, 2006; Romling, 2015) were selected because they are replicated in the genome of S. enterica several times (McClelland et al., 2001). For the third set, genes were used that had been incorporated into various methods such as MLST (Fakhr et al., 2005; Alcaine et al., 2006; Tankouo-Sandjong et al., 2007; Han et al., 2010; Bell et al., 2011; Stepan et al., 2011; Seong et al., 2012; Fresno et al., 2014), as housekeeping genes in transcription assays (Csonka et al., 1994; Galitski and Roth, 1997; Hensel et al., 1999; Tedin and Norel, 2001; Olson et al., 2007; Gilberthorpe and Poole, 2008; Malcova et al., 2009; Chan et al., 2012), or that were found to be part of a set of naturally mutated genes in S. enterica (Guard et al., 2011). Geneious 8.1.6 software was used to conduct BLAST searches, multiple alignments, and translations. After conducting BLAST searches, the parameters for keeping a hit for further analysis included being within S. enterica subspecies I, showing no indication of truncation, and having an appropriate gene length.
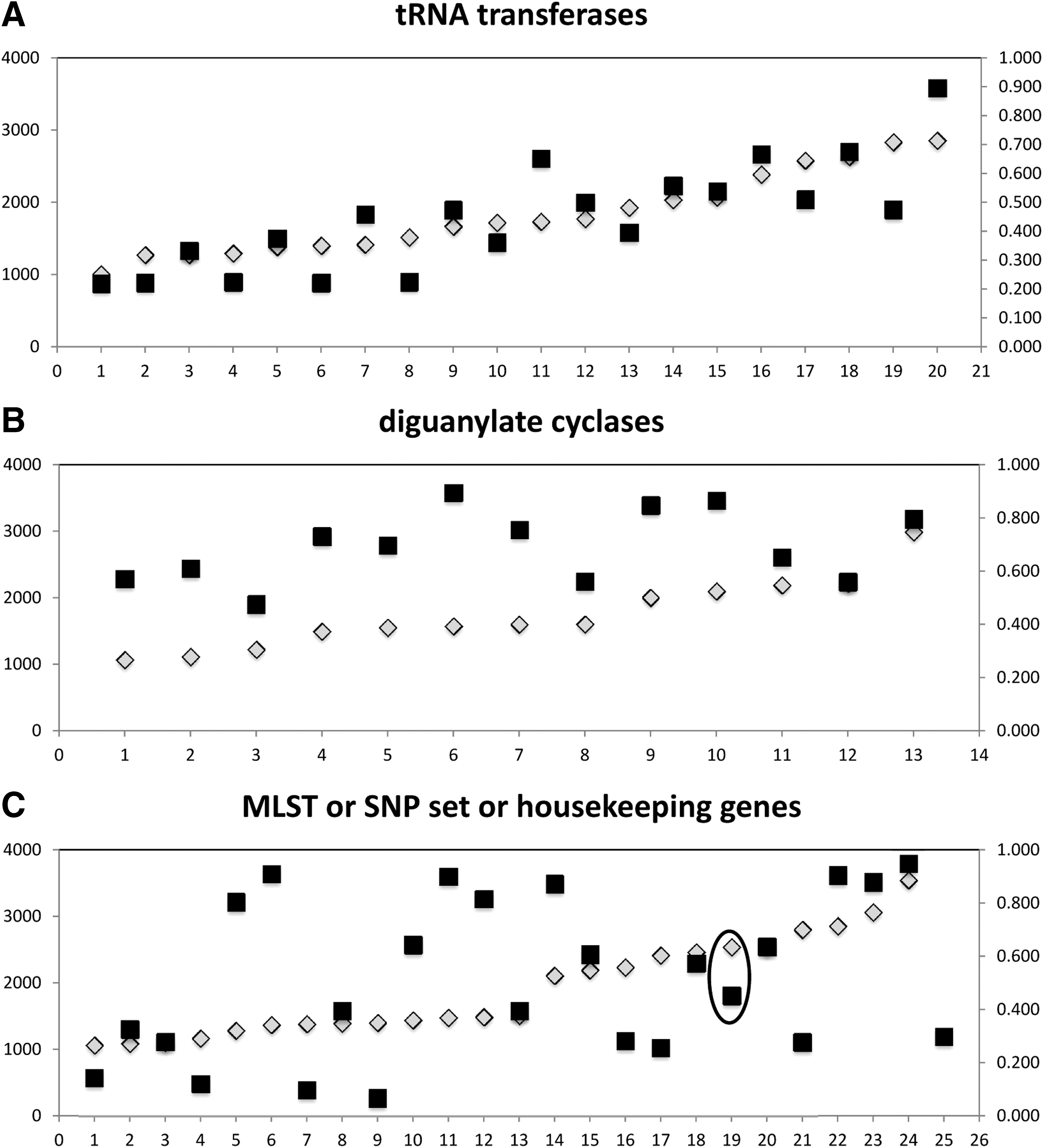
Determination of mutation indices for different classes of genes of Salmonella enterica subspecies I by metadata analysis of all available sequences at NCBI. Dates of data acquisition spanned from August to September of 2015. Gene identity correlating to numbers listed on the x axes and other information about genes are listed in Table 1. y axis (left, gray triangles), gene length in base pairs (gray triangles). y axis (right, black squares), calculated values for nonsynonymous/total SNPs per gene (NS/T mutation index). Graphing results are shown for 1
Reference genome was Salmonella enterica subspecies I serovar Typhimurium strain LT2 unless otherwise noted (NC_003197.1) (McClelland et al., 2001).
BLAST was done using Geneious software interface with NCBI to retrieve a minimum of 500 sequences; only S. enterica subspecies I sequences were included for analysis, and only if it had a complete gene. The multiple align function was used to identify unique DNA sequences. Sequences were translated and then analyzed again by the multiple align algorithm to identify the number of unique protein sequences.
As obtained from NCBI.
DGC, diguanylate cyclases; MLST, multilocus sequence typing; NCBI, National Center for Biotechnology Information; NS/T, nonsynonymous to total.
S. enterica strains used for ASPE and microsphere-based fluidics analysis of SNPs
The five reference S. enterica strains used for initial development were Enteritidis PT4 22079, Enteritidis PT13a 21046 (PT13a-wt), Enteritidis 13a 21027 (PT13a-bf), Typhimurium LT2, and Kentucky CDC191 (Guard et al., 2012). These strains and the other 25 S. enterica poultry-associated isolates shown in Table 2 were maintained according to previously described protocols (Guard et al., 2012). Of the serovars that were poultry associated and used to validate cyaA-targeted ASPE, serovars Typhimurium, Enteritidis, Newport, and Heidelberg are the first through fourth most common isolated from people (CDC-NCZEID, 2013). Serovars Montevideo, Schwarzengrund, and Agona often cause disease in people and have respective rankings of 7th, 23rd, and 30th (CDC-NCZEID, 2013). Serotype Kentucky is at most an infrequent cause of human disease, but there is concern that it carries a transmissible plasmid encoding antibiotic resistance (Le Hello et al., 2013). Serovars Gallinarum and Pullorum do not cause human disease, but they are closely related to serovar Enteritidis and are important avian pathogens that are subject to regulatory activities (Feng et al., 2013). Together, the isolates that were recovered in association with poultry and used here to validate ASPI cover a broad range of serovars and pathotypes.
Serovars Heidelberg and Typhimurium were differentiated by SNPs in the ISR region.
ISR, intergenic sequence ribotyping; SNP, single-nucleotide polymorphism.
Cell cultures were revived from frozen stock on brilliant green agar (Neogen, Lansing, MI) overnight at 37°C. One colony was transferred to brain-heart infusion broth (Neogen) and grown overnight at 37°C with shaking at 150 rpm. DNA was isolated from 1 mL of culture diluted to an optical density of 1.0 at wavelength = 600 nm with the PureLink Genomic DNA Mini Kit (Invitrogen, Grand Island, NY) following the manufacturer's instructions, including the RNase digestion step. Genomic DNA, 50 ng, was used as the template for the initial cyaA PCR. The cyaA PCR, ASPE hybridization, and SNP detection protocols were used as described for subtyping of all S. enterica isolates.
PCR assay design for gene cyaA
Entire cyaA genes (∼3 Kb) for the five S. enterica reference strains were retrieved from GenBank (Benson et al., 2013) and aligned using the MEGA 5.0 software package (Tamura et al., 2011). The resultant alignment file was truncated to a 300-basepairs (bp) region (bp 1900–2199 in STM3939) that was found to contain descriptive SNPs. General PCR cyaA primers were designed against conserved regions among all five reference strains: SAL-cyaA-F1 (5′-CCGGATAGCGTGGAGGTGTT-3′) and SAL-cyaA-R2 (5′-CACCACTGACGGCAATTTCACC-3′). The thermocycler used was a Realplex 4S (Eppendorf, Hauppauge, NY). The optimized cyaA PCR consisted of 50 ng DNA, AccuStart PCR 2× ToughMix (Quanta Biosciences, Gaithersburg, MD), and 400 nM each SAL-cya-F1 and SAL-cya-R2 primers (Biosearch, Novato, CA). The PCR program was 95°C for 10 min; 35 cycles of 95°C for 20 s, 60.6°C for 30 s, 72°C for 1 min; 72°C for 10 min.
To determine the specificity of the cyaA PCR assay to S. enterica, genomic DNA was extracted from a panel of negative controls (Campylobacter lari strain 43675, Campylobacter coli strain 33559, Campylobacter jejuni strain 14544, Escherichia coli strain EDL 933, and L. monocytogenes strain Li23). To determine the limit of detection for each reference strain, cyaA amplicons were generated using 0.001 pg–50 ng of template genomic DNA. For both the specificity and limit of detection tests, the cyaA PCR protocol described above and the hybridization and detection protocols described below were used as described.
ASPE primer design, cyaA amplicon hybridization, and SNP detection
The initial cyaA PCR assay used final primer concentrations = 400 nM, Tm = 60.6°C to produce amplicons for the ASPE reaction at genomic DNA template concentrations ≥0.001 ng per cyaA PCR. The ASPE primers (Table 3) were designed to contain both the reverse complement of the ANTI-TAG sequence attached to the MagPlex-TAG-coupled microspheres (Luminex), and a cyaA gene sequence with the 3′ terminal end representing the discriminatory SNP. Primers were designed for both the SNP and wild-type sequence for the reference S. enterica strains, and each primer was coupled to a unique MagPlex-TAG-coupled microsphere to perform multiplexed ASPE.
Bold portion of the primer represents the TAG sequence for the primer to hybridize to the microsphere, while the nonbolded portion represents the allelic portion ending in the discriminatory SNP (underlined base).
ASPE, allele-specific primer extension; SNP, single-nucleotide polymorphism.
Using the cyaA amplicons from the initial PCR step, 5 μL of that PCR was mixed with 2 μL ExoSAP-IT reagent (Affymetrix, Santa Clara, CA) and incubated at 37°C for 30 min, then 80°C for 15 min. The ASPE reaction was performed using 0.75 U Tsp DNA polymerase, ASPE reaction buffer (20 mM Tris-HCl, pH 8.4, 50 mM KCl), 1.25 mM MgCl2, 5 μM each of dATP, dTTP, dGTP, 5 μM biotin-14-dCTP, 25 nM each TAG-ASPE primer, and 5 μL ExoSAP IT-treated PCR template amplicon. All reagents used were from Invitrogen. The ASPE cycling program consisted of 96°C for 2 min; 30 cycles of 94°C for 30 s, 55°C for 1 min, 74°C for 2 min.
MagPlex-TAG-coupled microspheres were supplied by the manufacturer at a concentration of 2.5 × 106 microspheres/mL. A hybridization mixture was made of 1 μL of each of the eight MagPlex-TAG-coupled microspheres (equal to 2500 microspheres of each), 17 μL 2× Tm Hybridization Buffer (0.2 M Tris-HCl, pH 8.0, 0.4 M NaCl, 0.16% Triton X-100), 5 μL ASPE reaction, and 20 μL PCR water for a final volume of 50 μL. The hybridization was carried out at 96°C for 90 s and then 37°C for 30 min. The hybridized microspheres were pelleted by a magnetic separator (Perkin Elmer, Shelton, CT), washed with Tm Hybridization Buffer, and incubated with streptavidin-R-phycoerythrin (Invitrogen) at a final concentration of 2 μg/mL in 75 μL Tm Hybridization Buffer at 37°C for 15 min. Fifty microliters were analyzed at 37°C in the MAGPIX instrument (Luminex).
Sample data were analyzed using the xPONENT® ver. 4.2 software package (Luminex), and positive and negative signals for each microsphere bead pair (SNP and wild type) were determined in the following manner. The average median fluorescence intensity (MFI) of the two no template control (NTC) wells was subtracted from the MFI of a sample to yield the net MFI for each sample well. For each bead pair, a net MFI from the non-SNP-associated bead that was at least 30% higher than the SNP-associated bead indicated a negative signal for that SNP. Conversely, 30% higher from the SNP bead than the non-SNP bead indicated a positive for the SNP. A background sample consisted of a NTC from the PCR that had undergone ASPE and hybridization.
Validation of cyaA-targeted ASPE for analyzing S. enterica
The set of five Salmonella reference strains was tested in various combinations to determine the specificity of the method. Genomic DNA of a single strain was used as input to the initial PCR, followed by ASPE with one set of primers (one set of two primers, one primer for each allele) and hybridization with the two corresponding MagPlex-TAG-coupled microspheres; each strain was tested in this manner. First, 50 ng of genomic DNA of a single strain was used as input to the initial PCR, followed by ASPE with every possible combination of the four primer sets (two to four sets in various combinations) and hybridization with the corresponding MagPlex-TAG-coupled microspheres; each strain was tested in this manner. Finally, genomic DNA from the reference strains was mixed in every possible combination of two to five strains. For these combinations, two series were done: in one, the initial input to the PCR was 50 ng of each strain, regardless of the number of strains, and in another, the total input was 50 ng (two strains at 25 ng/strain, three strains at 16.7 ng/strain, four strains at 12.5 ng/strain, or five strains at 10 ng/strain). The resulting PCR amplicons from these combinations were used in ASPE reactions with all four primer sets and hybridization with all eight MagPlex-TAG-coupled microspheres. The panel of 25 S. enterica isolates encompassing 12 serotypes was analyzed using the optimized cyaA SNP method (Table 2). Genomic DNA was extracted from these isolates as explained before in the PCR Assay Design for Gene cyaA section.
Results
Review of cyaA SNPs present in the NCBI database
BLAST search using sequence from Salmonella enterica serotype Typhimurium LT2 (NC_003197.1) cyaA gene STM3939 recovered 378 sequences of cyaA from NCBI (Line 64 in S1). The set of cyaA sequences available was heavily weighted toward two serotypes. Of the 378 sequences in the set as of August 2015, 175 (46.3%) were from serotype Enteritidis and 116 (30.7%) were from serotype Paratyphi A. Other serotypes with more than three entries included serotypes Typhimurium with 21 (5.6%), Newport with 12 (3.2%), and Heidelberg with 5 (1.3%). All other serotypes had fewer than five representative strains. Percent GC content of cyaA within S. enterica subspecies I ranged from 54.2% to 54.7%, percent identical sites ranged from 98.6% to 100%, and reported gene lengths were from 2543 to 2547 bp. Gene length depended on inclusion of terminating codons, and thus, all sequences were trimmed to a length of 2540 bp. Among the 378 sequences meeting parameters of the cyaA BLAST, 42 DNA sequences were unique, and these translated into 19 unique amino acid sequences. The NS/T change thus gave a mutation index of 19/42 or 0.452 (Fig. 1). In other words, a little less than one amino acid was altered for approximately every two differences in nucleotides.
To compare the NS/T of cyaA to other genes, a set of 20 tRNA transferases greater than 1000 bp was analyzed, and there was one gene analyzed per common amino acid. This set of genes was chosen because it was hypothesized to be highly evolved and thus likely to have a fairly stable NS/T mutation index. Figure 1A indicates that the NS/T index may be impacted by the size of the gene, so results are listed relative to increasing gene size. For tRNA transferases between 1000 and 2000 bp, the average NS/T index was 0.359 and the standard deviation was 0.1421. For genes between 2000 and 3000 bp, the average NS/T index was 0.617 and the standard deviation was 0.1444. Thus, NS/T indices for the tRNA transferases are an example of SNP variation increasing proportionately to gene size. The five tRNA transferases least likely to generate amino acid sequence variation were serS, asnC, lysS, leuS, and ileS, and the entire class had an average NS/T index of 0.449. The NS/T index for cyaA of 0.452 suggests that cyaA has an amino acid sequence about as stable as that of an average tRNA transferase.
Results from analysis of the DGC differed (Fig. 1B). For genes between 1000 and 2000 bp, the average NS/T index was 0.662 and the standard deviation was 0.1337, whereas respective values for genes greater than 2000 bp were 0.744 and 0.1329. Although NS/T indices for shorter versus longer genes for tRNA transferases were significantly different (p = 0.0009), the same parameter for the DGC genes was not (p = 0.3055). These results suggest that DGC genes of S. enterica subspecies I are significantly more likely to generate amino acid sequence variation than the tRNA transferases, regardless of gene size (p = 0.0003). All of the genes in the DGC class appear to undergo significantly frequent amino acid substitution in comparison to the tRNA transferases.
The third set of genes analyzed was chosen with no emphasis on relatedness of function, but they were used in other genomic investigations. The cyaA gene was included in this set (Fig. 1C, circled datapoint). Results indicate that NS/T indices did not differ significantly according to the length of the gene (p = 0.2907). The standard deviation in NS/T indices for this third set of genes was 0.299, whereas it was 0.185 and 0.134 for tRNA transferases and DGC genes, respectively. Twelve genes in set 3 had NS/T indices less than cyaA, and 12 genes had NS/T indices that were greater (Table 1). These results suggest that random selection of genes is likely to generate some variation between selected gene targets that is due to innate differences in mutation index. The gene cyaA appears to be located at a midpoint of variation.
Accuracy and sensitivity of cyaA SNP detection by ASPE for S. enterica strains
For the four SNP-containing reference strains (Enteritidis PT13a-wt, Enteritidis PT13a-bf, Typhimurium, Kentucky), all possible combinations (from singleplex to five-plex) yielded expected SNP patterns and always matched the actual patterns determined by the assay with 100% accuracy. The fifth strain, Enteritidis PT4, was used as a negative control since it does not possess an SNP within this 300-bp region of the cyaA gene. However, Table 4 shows the large number of SNPs that could detect Enteritidis PT4 and other phage types as a positive reaction in future assays. The 100% SNP pattern detection accuracy did not change based on the two different ways in which the template combinations were created (50 ng DNA for each strain or 50 ng DNA total). These results highlight the specificity and accuracy of cyaA SNP detection using this optimized assay against the reference strains used to develop the ASPE primers and MagPlex-TAG magnetic microspheres.
DNA nucleotide code: g, guanosine; a, adenosine; t, thymidine; c, cytidine; w, a or t, weak (two H-bonds); s, c or g, strong (three H-bonds); m, a or c, aMino; y, c or t, pYrimidine; r, g or a, puRine.
SNPs in bold are within the coding region for cyaA flanked by assay primers F1 and R1; the SNP in italics at bp 2019 differentiates phage type lineages of serovar Enteritidis; the PT4 lineage (NC_011294) has a C, PT13a/8/14b lineage has a T (NZ_CP007175).
An unlisted SNP at bp 2105 further distinguishes serovar Enteritidis wild-type PT13a and PT4 from a biofilm forming strain of PT13a. The first two have an adenosine (a) and the latter has a cytidine (c) (Morales et al., 2007; Guard et al., 2011).
SNP, single-nucleotide polymorphism.
Validation of ASPE for poultry-related Salmonella enterica serotype Enteritidis environmental isolates
The cyaA SNP assay demonstrated high agreement between the expected and actual SNP pattern observed for the different serotypes within the environmental isolate panel (89%; Table 2). It was expected that serotype Heidelberg would not be differentiated from serovar Typhimurium, because it lacked an SNP in the region under investigation. As expected, poultry-related serotypes not used to design the assay (Heidelberg, Agona, Cerro, Gallinarum, Infantis, Montevideo, Newport, Pullorum, Schwarzengrund) did not have SNPs for any of the target bases. However, review of available cyaA sequences revealed that many other SNPs could be used to target other serotypes (Table 4). Two pathotypes of Phage Type 13a serotype Enteritidis, namely the egg-contaminating strain (PT13a-wt 21046) and a biofilm-forming non-egg contaminating strain (PT13a-bf 21027), had discriminatory SNPs in the cyaA gene (Morales et al., 2007). When used to group a panel of poultry-related environmental Enteritidis isolates, the correct SNP pattern was found for the two known subtypes of PT13a. PT13a and PT8 belong in the same evolutionary lineage and vary by plasmid content, so they would be expected to group together (Threlfall et al., 1993; Liebana et al., 2004). One PT14b grouped with the PT13a wt strain 21046, which is a finding supported as correct by results from NCBI bioproject 219482 (Rehman et al., 2014). As expected, two PT4 isolates did not have either of the SNPs associated with the PT13a/PT8 lineage (Thomson et al., 2008). These results indicated that the cyaA SNP assay worked well for distinguishing between previously characterized PT13a pathotypes and could distinguish the PT4 lineage from PT13a/8.
Conclusions
These analyses suggest that cyaA SNPs targeted by ASPE will support and extend the use of ISR applied as a screening method for assigning serotype to S. enterica. It will help distinguish between serovars that might share the same ISR sequence (e.g., ISR group UN0006), provide some information about phage type and pathotype, and ultimately achieve some subtyping within serotype. An incidental finding is that tRNA transferases and cyaA give a conservative assessment of subtype in comparison to DGC genes. Thus, DGC genes might be most valuable for identifying strains rapidly emerging within subtypes even within a single outbreak. Given the ability of the Luminex MagPlex system to identify up to 150 custom beads within a single well, assays can be developed and optimized to detect up to 75 different SNPs. Thus, finding at least 84 SNPs across a gene that is 2540 bp suggests that cyaA is an ideal target for assay development. Cost per sample for bead-based capillary systems has been quoted to range from $40 to $50 per sample, but the ability to process multiple SNPs within single wells might make it an affordable confirmatory assay for properly equipped laboratories. Other methods that do not require specialized equipment or intensive maintenance regimens, such as ISR, appear less costly for conducting routine screening for serotype and field investigations of on-farm ecology (Guard et al., 2012; Jean-Gilles Beaubrun et al., 2014).
Comparative whole genome analyses have revealed a large number of potentially discriminatory SNPs among many different genes (Zheng et al., 2014). In this instance, SNP analysis was done within the context of how likely cyaA is to mutate compared to different sets of genes, and it was assessed for the number of target sites it has that are amenable to analysis by xMAP technology. As information is assessed by whole genome analysis from hundreds of strains, target sites will be identified that provide optimal genomic information about sources of outbreaks that will facilitate protecting the safety of the food supply.
Footnotes
Acknowledgments
The authors would like to acknowledge the expert technical assistance of Laura Lee-Rutherford in the development of primer sets and optimization of SNP assays, and Tod Stewart for assistance with the operation and maintenance of the MagPix System. These investigations were supported through the U.S. Poultry and Egg Association Foundation Project No. F043 as well as additional support from Agricultural Research Service, USDA CRIS Projects “Pathogen Reduction and Processing Parameters in Poultry Processing Systems” No. 6612-41420-017-00 and “Genetic Analysis of Poultry-Associated Salmonella enterica to Identify and Characterize Properties and Markers Associated with Egg-Borne Transmission of Illness” No. 6612-32000-007-00.
Authors' Contributions
J.G. designed and implemented initial research, M.J.R. designed, developed, and conducted fluids experimentation, and J.G. and M.J.R. wrote the article. Z.A., S.O.B., and P.K. provided statistical oversight and some bioinformatics support.
Disclosure Statement
No competing financial interests exist.