
Abstract
To study the relationship between environmental and clinical populations of Vibrio parahaemolyticus, we collected in total 86 isolates from Southern China during one and a half years. Sixty-eight isolates were recovered from aquaculture ponds, a seafood market, and restaurants, and 18 isolates were recovered from clinical samples. Virulence gene analysis revealed that 25 isolates (14 clinical and 11 environmental) tested positive for tdh, but only 4 carried trh. Interestingly, none of the tdh + environmental isolates was recovered from ponds. Both environmental and clinical tdh+ isolates, except for one clinical isolate, harbor type III secretion system 2α (T3SS2α) and T3SS2β-related genes, including vopB2α, which was previously suggested to be absent from environmental strains. More than 70% of clinical isolates carried the pandemic marker of new toxRS (GS-PCR+), which was not present in the environmental isolates. Pulsed-field gel electrophoresis and multilocus sequence typing analysis showed a high degree of genetic diversity within the environmental isolates. In contrast, the clinical population formed a tight cluster that differed from the environmental isolates. These findings suggest that the pandemic strains of V. parahaemolyticus may not directly originate from marine animals. Rather the environments where they are maintained could serve as reservoirs for toxigenic, but not pandemic strains. These environments provide an ideal place for generation of new toxigenic strains through DNA exchange, which was revealed by extensive recombination events in recA sequences of the environmental isolates.
Introduction
V
Virulence factors of V. parahaemolyticus have been extensively studied. These factors include various toxins and effectors of the type III secretion system (T3SS) (Nishibuchi and Kaper, 1995; Izutsu et al., 2008). Thermostable direct hemolysin (TDH) and TDH-related hemolysin are two well-characterized V. parahaemolyticus toxins. Strains that are tdh positive show a hemolytic activity on Wagatsuma agar, which is an important clinical marker to identify pathogenic strains (Honda and Iida, 1993). In previous epidemiological studies, tdh and trh were detected in 99% of clinical strains, but only in 2–3% of environmental strains (Nishibuchi and Kaper, 1995; Roque et al., 2009). T3SSs use a needle-like structure to deliver bacterial effectors directly into host cells (Ham and Orth, 2012). V. parahaemolyticus possesses two T3SS systems that are located on the chromosome 1 and 2, respectively (Makino et al., 2003). T3SS1 has high homologies with that in other vibrio species. T3SS2 is only present in pathogenic strains of Vibrio cholerae and V. parahaemolyticus (Dziejman et al., 2005). Two distinct lineages of T3SS2, T3SS2α, and T3SS2β have been described for V. parahaemolyticus. Presence of tdh correlates with T3SS2α, whereas presence of trh correlates with T3SS2β (Park et al., 2004; Okada et al., 2009).
In China and South East Asia, people prefer to consume fresh instead of frozen seafood. Consequently, aquatic animals are maintained alive shortly before consumption. Clinical strains are consequently considered to have been transmitted through marine animals (Chao et al., 2011). However, there are no data available that describe the relationship among strains recovered from the aquaculture ponds, temporary holding environments, and strains from clinical samples. In this study, we surveyed and isolated V. parahaemolyticus from different consumer food chain environments associated with Southern China, including aquaculture ponds, a seafood market, restaurants, and clinics during a time period of one and a half years. Virulence traits, genetic diversity, and population structure of the isolates were analyzed by multiple approaches. Our results may shed light on the origin and transmission of pathogenic strains of V. parahaemolyticus from marine environment to humans.
Materials and Methods
Sample collection
Samples were collected from three aquaculture ponds, a seafood wholesale market, two restaurants, and four hospitals located in Hainan and Guangdong Provinces during 2010–2012 and are recorded in Supplementary Table S1 (Supplementary Data are available online at
Identification of bacterial strains
Bacteria isolated from the samples were subcultured on thiosulfate citrate bile salt sucrose agar (BD Diagnostics). Single green colonies were picked and cultured on TSA with addition of 2% NaCl. Bacterial DNA was obtained with an Axyprep Bacterial Genomic DNA Miniprep Kit (Axygen Scientific) and used as template for amplification of 16S rDNA genes and gyrB. Nucleotide sequences of both genes were sequenced and then run through a megablast search to identify the highly similar sequences (
Detection of virulence genetic markers
Fifteen sets of oligonucleotide primers (Table 1) were designed to detect the presence of virulence genes as well as the pandemic marker (toxRS). Polymerase chain reaction (PCR) cycles were run as described in Table 1. Amplified products were separated by electrophoresis in 1% agarose gel. DNA from E379 (ATCC33843) and E154 (ATCC17802) that are positive for all the virulence markers served as a positive control and DNA from Escherichia coli DH5α served as negative control. To avoid false-positive reaction, products were purified and sequenced to confirm identity of the respective gene.
Molecular typing of V. parahaemolyticus by pulsed-field gel electrophoresis
Pulsed-field gel electrophoresis (PFGE) analysis was performed according to the standard protocol of PulseNet (
Multilocus sequence typing analysis
Multiple sequence alignments (MSAs) for each locus (gyrB, recA, toxR) were aligned and trimmed by Clustal W in MEGA 5 (Tamura et al., 2011). The in-frame sequences at the three loci were concatenated to produce one sequence. A phylogenetic tree was constructed from the allelic profile with the unweighted-pair group method (UPGMA) using average linkages. The tree was subsequently created in MEGA5. Numbers of polymorphic sites, the parameters Pi and dN/dS, as well as the haplotypes were determined for individual loci and concatenated sequences with DnaSP 5 (Librado and Rozas, 2009). Recombination events were detected using the Recombination Detection Program version 3.0 (RDP3) (Martin et al., 2005). The structural genetic relationship between each population was determined with Arlequin3 following the instructions described by Excoffier et al. (2007).
Results
Purification and detection of V. parahaemolyticus
Over 900 isolates were collected during one and a half years. The detection rates of V. parahaemolyticus differed depending on the sampling site. From ponds, the market, and restaurants, 3.4%, 4.41%, and 12.34% of all isolates were identified as V. parahaemolyticus. Eighty-six isolates and two reference strains were included in the genetic analysis. They were assigned to four groups based on their origin: aquaculture ponds (12.5%; 11 isolates), seafood market (20.5%; 18 isolates), restaurants (44.3%; 39 isolates), and clinics (20.5%; 18 isolates).
Virulence traits of the V. parahaemolyticus isolates
The isolates were screened for 15 virulence markers, including genes encoding for hemolysins, T3SS1, T3SS2α, and T3SS2β and the pandemic marker of group-specific PCR (GS-PCR). The major virulence gene tdh was detected in 14 clinical strains (77.8%) and 11 environmental isolates (16.2%), of which 6 came from the market (33.3%), 5 came from the restaurants (13.2%), but none from the ponds. The gene tlh was present in all isolates, except one from the ponds. The trh gene was present in only four isolates (4.5%), of which two were from the environment and the other two from clinical samples. Three of these trh-positive isolates also carried tdh (Table 2).
GS-PCR, group-specific polymerase chain reaction; T3SS, type III secretion system.
To detect presence of T3SS1, four genes, vpa0450, vopS, vopR, and vopQ were used to screen isolates. All isolates, except one from the market possessed at least one of four genes (Table 2). 89.5% of the isolates harbored vpa0450, 97.7% harbored vopS, 93.0% harbored vopR, and 91.9% harbored vopQ. To detect presence of T3SS2α five genes, vopB2α (VP1362), vopL, vopA, vopT, and vopC were used to screen 86 isolates. Of the 68 environmental isolates, the tdh+/trh− isolates tested positive for all five genes, including vopB2α (VP1362), which could not be detected in previous studies (Jones et al., 2012). Two tdh−/trh− environmental isolates (DDC05 and HS43-2) carried part of the T3SS2α genes. In the remaining environmental isolates, none of the T3SS2α genes were present. Of the clinical samples, all tdh+/trh− isolates contained the five genes, whereas all of the tdh−/trh− isolates contained at least two of these genes, including vopB2α. One tdh+ /trh+ isolate did not harbor any of T3SS2α gene. To detect presence of T3SS2β genes, primers for vopB2β and vopD2β were employed. 74.4% carried vopB2β and 97.6% carried vopD2β, of which only four isolates contained trh. To further screen the isolates, a GS-PCR was used, which targets a new toxRS region that is associated with pandemic strains (tdh+ /trh− ). None of the environmental isolates were positive for this marker. However, in 13 of the 18 clinical isolates, this region was detected. Of these 13 isolates, 12 were tdh+ / trh− and 1 isolate ESS12070 was tdh−/trh− , but had previously been tested positive for four genes of T3SS2α (Table 2).
Molecular typing of V. parahaemolyticus by PFGE
PFGE analysis showed a high level of diversity among 86 isolates. Sixty-eight distinct patterns were distinguished according to the criteria suggested by Tenover et al. (1995), where two band profiles are considered to be the same pattern if their difference is not greater than three bands (Fig. 1 and Supplementary Table S1). Pattern P29 was the dominant profile and contained seven isolates, of which all were isolated from ponds and the market (Supplementary Table S1). The tdh+ strains were characterized by PFGE patterns (Fig. 2). Five groups were distinguished at a similarity cutoff level of 80%. Group I with three isolates was recovered from the same patient in 2011. However, the isolates were not identical concerning virulence markers (Table 3). Group II with four isolates originated from four different patients. Group III contained three isolates, which were either recovered from the market or restaurants. Group IV contained four strains that had been isolated from four different patients. Group V encompasses four isolates that originated from the seafood market and were sampled on the same day. Interestingly, except group I, the isolates within each group had identical virulence markers (Table 3).

PFGE patterns of NotI-digested genomic DNA of Vibrio parahaemolyticus isolates. Isolates were classified into 68 distinct patterns. Two profiles were considered to be the same if the difference did not exceed three bands. PFGE, pulsed-field gel electrophoresis.
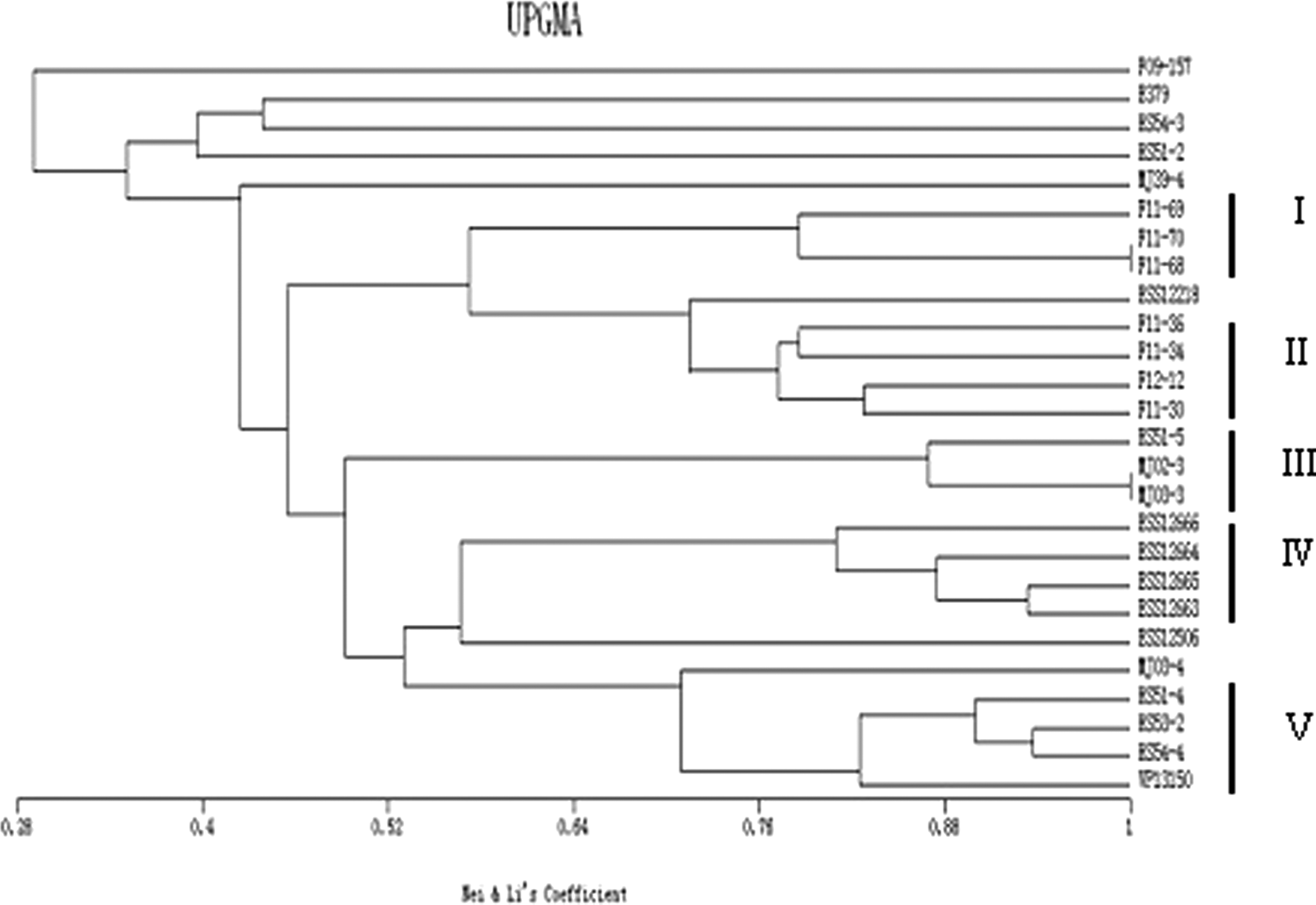
PFGE pattern of the tdh+ isolates clustered with the UPGMA method. Five groups were distinguished at a similarity cutoff level of 80%. Group I, II, and IV were composed of clinical isolates, but group III and V were composed of environmental isolates. PFGE, pulsed-field gel electrophoresis; UPGMA, unweighted-pair group method.
+, positive amplification; −, negative amplification; E, environmental; C, clinical.
GS-PCR, group-specific polymerase chain reaction; PFGE, pulsed-field gel electrophoresis; T3SS, type III secretion system.
Multilocus sequence typing analysis of V. parahaemolyticus
MSAs for each locus (gyrB, recA, toxR) were aligned and trimmed by Clustal W in MEGA 5. A majority consensus phylogeny was constructed from concatenated loci (1794 bp). Twelve clades were identified in the dendrogram as shown in Figure 3. Clade 1 comprised nine clinical isolates that were all tdh+ , except for ESS12070. Interestingly, these strains were characterized as pandemic strains that carry the GS-PCR marker (Supplementary Table S1). Clade 7 consists of nine isolates, of which all are tdh+ , but had been isolated from the environment. Compared to clade 1, they lacked the GS-PCR marker. The rest of clades contained only environmental strains. None of the clades contained both environmental and clinical isolates.
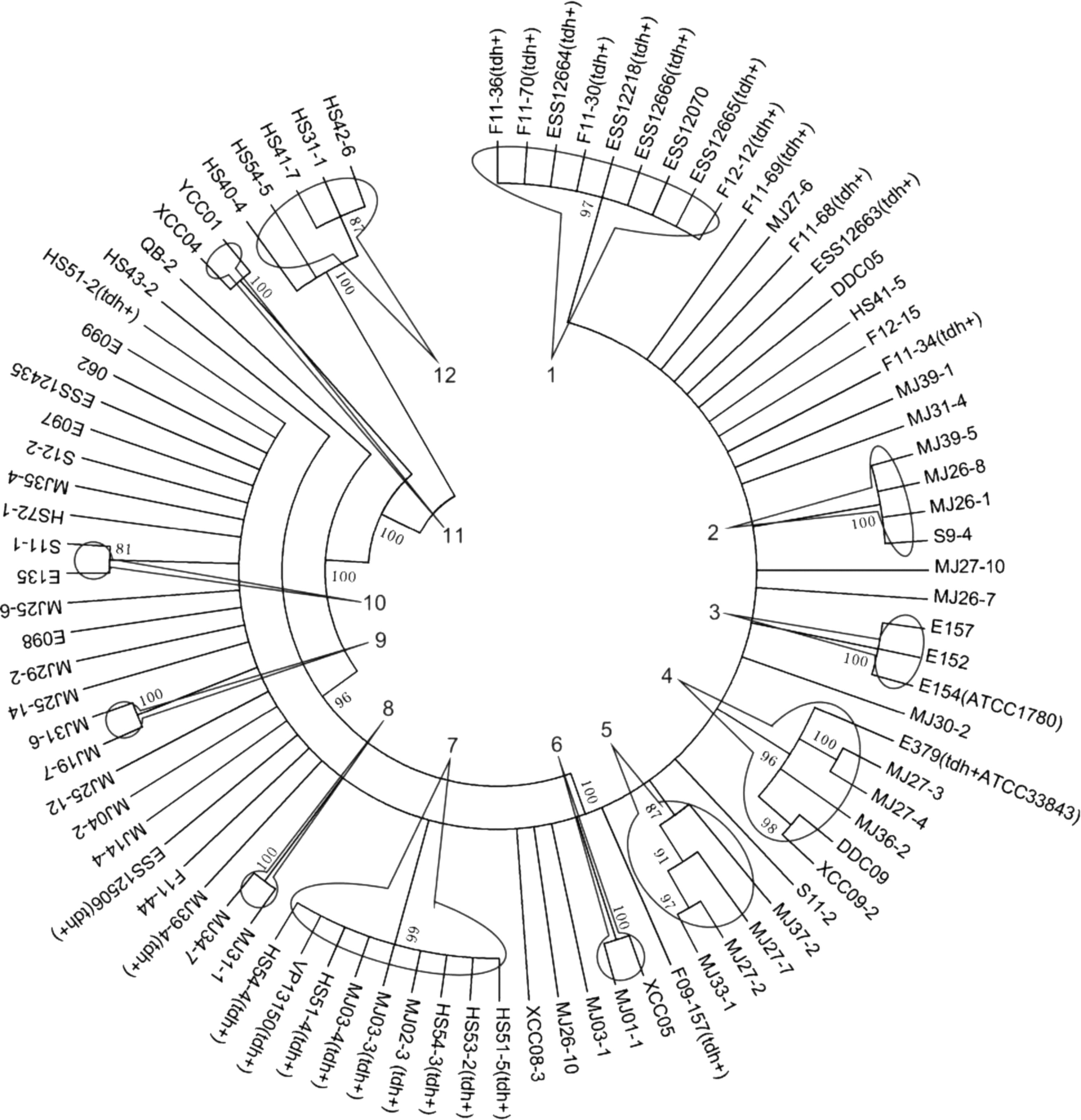
Phylogenetic tree derived from concatenated sequences of gyrB, recA, and toxR of 86 V. parahaemolyticus constructed by the UPGMA method. Clustering was performed at similarities of 80% and yielded 12 clusters. UPGMA, unweighted-pair group method.
The genetic diversity of isolates was analyzed with Dnsp5.0. As shown in Table 4, internal fragment sizes of gyrB, recA, and toxR are 406, 776, and 615 bp, respectively. Numbers of alleles are 33, 42, and 36. Polymorphic sites for each locus are 27, 257, and 42. Average nucleotide diversities (pi) are 0.0129, 0.0535, and 0.0096. The rates of nonsynonymous substitutions range from 0.0007 (gyrB) to 0.013 (recA) and the synonymous substitutions range from 0.027 (toxR) to 0.183 (recA). The ratio of nonsynonymous to synonymous mutations show that all loci were subjected to purifying selection since the dN/dS ratios are <1 for each locus.
A total of 61 haplotypes were detected among the concatenated sequences of 86 strains (Supplementary Table S2). With respect to epidemiological sources, 9 haplotypes were found in the pond (n = 11), 13 in the market (n = 18), 28 in the restaurant (n = 39), and 10 in the clinical samples (n = 18). Hap_15 encompassed environmental tdh + isolates that are found in Clade 7. Hap_54 encompassed clinical tdh + isolates that are found in Clade 1. The genetic structures of the four populations were compared by Alequein3 based on the haplotype frequencies. The results showed that the V. parahaemolyticus population associated with the aquaculture ponds was not genetically related to the populations associated with other sites. The same was true for the clinical isolates (p < 0.05), but the genetic differences between the isolates associated with the market and restaurant populations were not significant (p = 0.075) (Table 5). These results suggest that these food chain sites are sites where acquisition of new virulence factor genes may be occurring.
Significant difference, p < 0.05.
The recombination events of the gyrB, recA, and toxR and their concatenated sequences were examined. We could not detect recombination events in gyrB and toxR. However, 78 and 142 recombination signals with 2 and 11 unique recombination events were found in recA and the concatenated sequences, respectively. Three highly divergent alleles, recA-QB2, recA-XCC04, and recA-HS31, were identified in eight environmental isolates that with nucleotide Blast best matched counterparts in Vibrio campbelli, Photobacterium damselae, and Shewanella sp., respectively. Likely, those isolates have undergone interspecies DNA exchange involving entire fragments of recA. In addition, various mosaic structures of recA that are caused by intragenic recombination were found in 44 isolates. Interestingly, in 52 isolates with recombinant recA, only three belonged to clinical group and none of them were pandemic isolates (Supplementary Fig. S1).
Discussion
In this study, 68 isolates were recovered from different environments. In the market and restaurant, V. parahaemolyticus was highly prevalent. 19.3% of these isolates were found to carry tdh and other virulence markers. These data suggest that holding conditions for aquatic animals at the market and restaurants are at high risk for V. parahaemolyticus contamination and for consumers. In our investigation, 16.2% of the environmental isolates carried tdh gene, which seems much higher than other reports (Nishibuchi and Kaper, 1995; DePaola et al., 2003). However, in this study, all of the tdh+ strains were isolated from the market and restaurants, but not from the ponds, which suggests that toxigenic strains occur at a low rate in aquaculture environments. As a matter of fact, previous studies have reported that occurrences of potential pathogenic strains from fish markets and restaurants range from 8% to 15% (Alam et al., 2009; Duan et al., 2011; Jun et al., 2012), whereas the range is 1–2.5% in the aquaculture environment (Alam et al., 2002; Cook et al., 2002; Wagley et al., 2009).
Interestingly, the distribution of the tdh and trh genes in the pathogenic strains seems to be related to the geographic regions. In our investigation the predominant pathogenic strains, clinical or environmental, were tdh+ and trh− , which is in accordance with findings from previous studies in Southeast Asia and South America, where the pandemic strains (tdh +/trh− ) are more frequently distributed (Laohaprertthisan et al., 2003; García et al., 2009; Zhao et al., 2011). However, in Europe, where cases of infection by V. parahaemolyticus have rarely been reported, most of the pathogenic strains were tdh− or trh + (Robert-Pillot et al., 2004; Ellingsen et al., 2008; Ottaviani et al., 2013). Most potential toxigenic isolates from the U.S. Pacific coast and Gulf of Mexico have both tdh and trh genes (DePaola et al., 2000; Paranjpye et al., 2012).
T3SS1 was present in almost all of our V. parahaemolyticus isolates, regardless of their origin. Presence of T3SS2α was generally associated with tdh gene, which is in agreement with many reports (Meador et al., 2007; Han et al., 2008). In contrast to the observation by Noriea et al. (2010) and Jones et al. (2012), we found vopB2α (T3SS2α) in both environmental and clinical tdh+ isolates. This divergence might be due to genetic differences between populations from two geographic areas or sequence variation. Nevertheless, it is clear that diversity of V. parahaemolyticus is still beyond present understanding and thus, it is controversial to utilize vopB2α as a specific indicator of strains virulence as suggested by Noriea et al. (2008) and Jones et al. (2012).
Although environmental and clinical tdh+ strains carried similar virulence markers, they exhibited great variation with respect to genetic profiles. Above 70% of the clinical isolates were typical pandemic strains carrying the marker of GS-PCR, while none of the environmental isolates contained this marker, which is in line with previous reports (Noriea et al., 2010; Yu et al., 2013). Moreover, the tdh+ strains were clustered into distinct environmental and clinical groups based on PFGE or multilocus sequence typing profiles. These differences indicate that pandemic strains may not arise directly from environmental strains. This hypothesis is supported by previous studies. Yan et al. (2011) suggested that V. parahaemolyticus has evolved as a typical epidemic population with a different origin than the nonclinical isolates. Theethakaew et al. (2013) found that clinical samples formed a tight cluster different from the environmental population, and thus suggested that disease-causing isolates were not a random sample of the environmental reservoir.
Extensive recombination events in recA nucleotide sequences were found by previous studies as well as in this study (Chowdhury et al., 2004; González-Escalona et al., 2008; Yu et al., 2011). In contrast to the observation by Theethakaew et al. (2013), the predominant strains with the recombinant recA alleles were recovered from the environment and not from clinical samples. This could indicate that the human intestinal tract is not, as previously hypothesized, a hot spot driving the emergence of new potentially pathogenic strains (Theethakaew et al., 2013). It is unclear what causes this difference, and further studies are required to explore the origin of pathogenic and pandemic strains of V. parahaemolyticus.
Conclusion
This study has revealed that population structures of V. parahaemolyticus from aquaculture ponds, sea food markets, and restaurants and clinical cases were significantly different. Even though tdh + isolates from the environment carry a wide range of virulence markers, they are not related to clinical isolates, which suggests that the pandemic strains may not originate from fresh seafood or associated environments. A high rate of recombination events in the recA gene of the environmental strains suggests that V. parahaemolyticus may frequently change its genetic traits in the environment by horizontal gene transfer or by DNA rearrangement.
Footnotes
Acknowledgments
This work was supported by the Programme of Knowledge Innovation of CAS (KZCX2-EW-Q215), the National Natural Science Foundation of China (31272697), 12th five-year-major-projects of China's Ministry of Public Health (2012zx10004-213), the Canton-CSA United Foundation (2009B1300157), the National Natural Science Foundation of China (41276163), and the Project of Science and Technology New Star of Zhujiang in Guangzhou city (2013J2200094), which are acknowledged.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.