
Abstract
Cattle are a natural reservoir of Shiga toxin–producing Escherichia coli (STEC) and have recently been recognized as a major source of Campylobacter jejuni contamination. While several factors are known to be associated with bacterial colonization, the underlying microbial factors have not been clarified. In this study, we characterized the fecal microbiota of dairy cattle (n = 24) using next-generation sequencing to elucidate the intestinal bacterial communities and the microbial diversity in relation to the presence of the foodborne pathogens STEC and C. jejuni (STEC-positive samples, n = 9; STEC-negative samples, n = 15; C. jejuni-positive samples, n = 9; and C. jejuni-negative samples, n = 15). While no significant differences were observed in alpha diversity between STEC-positive and STEC-negative samples, a high diversity index was observed in C. jejuni-positive samples compared to C. jejuni-negative samples. Nine phyla, 13 classes, 18 orders, 47 families, 148 genera, and 261 species were found to be the core microbiota in dairy cattle, covering 80.0–100.0% of the fecal microbial community. Diverse microbial communities were observed between cattle shedding foodborne pathogens and nonshedding cattle. C. jejuni-positive cattle had a higher relative abundance of Bacteroidetes (p = 0.035) and a lower relative abundance of Firmicutes (p = 0.035) compared to C. jejuni-negative cattle. In addition, while the relative abundance of 2 and 6 genera was significantly higher in cattle-shedding STEC and C. jejuni, respectively, the relative abundance of 3 genera was lower in both STEC- and C. jejuni-negative cattle. Our findings provide fundamental information on the bacterial ecology in cattle feces and might be useful in developing strategies to reduce STEC or C. jejuni shedding in dairy cattle, thereby reducing the incidence of STEC infection and campylobacteriosis in humans.
Introduction
F
Research on the individual factors underlying the bacterial shedding of STEC or Campylobacter in cattle has been limited. Microbiota comprise an important individual factor playing a critical role in animal health, physiology, productivity, and bacterial shedding (Guarner and Malagelada, 2003; Chase-Topping et al., 2008). Indigenous microbes may inhibit or promote the colonization of pathogens by competing for nutrition or using the byproducts of indigenous bacteria (Poole et al., 2003; Stern et al., 2006). Organic acids and volatile fatty acids and the presence of butyrate-producing bacteria have been suggested to inhibit STEC shedding, and several bacterial species might promote or inhibit STEC shedding (Shin et al., 2002; Zhao et al., 2013). An increase in generic E. coli might inhibit Campylobacter colonization in mice (Haag et al., 2012). However, these studies involved culture-based techniques, limiting the understanding of microbial ecology in cattle.
Recent advancements in molecular methodologies enable the investigation of microbial communities regardless of the culture methods, facilitating the characterization of the entire bacterial population in a sample. The microbial diversity in relation to STEC or C. jejuni shedding has been reported, but not in dairy cattle (Durso et al., 2010; Rapp et al., 2012; Xu et al., 2014; Aluthge, 2015). Bacterial shift has been reported in Campylobacter-infected humans and mice (Haag et al., 2012; Dicksved et al., 2014), and differences in bacterial communities have been reported in Campylobacter-shedding chickens (Sofka et al., 2015). Moreover, while STEC infection may be asymptomatic in cattle, the presence of STEC might influence the composition of intestinal microbiota in beef cattle (Zhao et al., 2013; Xu et al., 2014). Limited information on this aspect is available for dairy cattle, the management of which differs from that of beef cattle, highlighting the necessity of microbial characterization in dairy cattle in relation to foodborne pathogen shedding. Furthermore, the influence of Campylobacter infection on cattle microbiota has not been investigated. Therefore, we characterized the fecal microbiota of dairy cattle using next-generation sequencing (NGS) to identify their intestinal bacterial communities and assess the microbial diversity in relation to the presence of STEC and C. jejuni.
Materials and Methods
Fecal sampling and cattle information
A single dairy cattle farm at Gyeonggi-do, South Korea, was visited regularly for sample collection from August 2012 to December 2014.
Feces were collected by rectal grab sampling. Approximately, 20 g of fresh feces was transported into a sterile specimen cup using a sterile spatula and immediately transported to the laboratory under 4°C. Some samples were subjected to bacterial isolation, while the remaining samples were stored at −70°C for microbial community analysis.
STEC and C. jejuni isolation
STEC O157 was isolated using the standard selective culture method and immunomagnetic separation (IMS) as described previously (Dong et al., 2014b). Briefly, 1 g of fecal sample was homogenized with 9 mL of modified EC broth (mEC; Becton, Dickinson and Company, Rutherford, NJ) supplemented with novobiocin (20 mg/L; Oxoid, Hampshire, UK) and incubated at 37°C overnight. Then, 1 loop of incubated mEC broth was subcultured on sorbitol MacConkey agar (Becton, Dickinson and Company) supplemented with potassium tellurite (T-SMAC, 2.5 mg/L; Sigma-Aldrich Canada Ltd., Oakville, ON, Canada) and incubated at 37°C overnight for the standard culture method. For the IMS method, 1 mL of enriched mEC broth was mixed with immunomagnetic beads coated with anti-E. coli O157 (Dynal; Invitrogen, Gaithersburg, MD), and the beads were separated using a magnetic bar according to the manufacturer's guidelines. The final suspension was spread onto T-SMAC and then incubated at 37°C overnight. The white colonies were then further tested on MacConkey agar, and the presumptive colonies in selective agar were tested for serotype and for the presence of Shiga toxin genes (stx1 and stx2) using the Latex Test Kit (Oxoid) and polymerase chain reaction (PCR) (Feng and Lampel, 1994), respectively. Non-O157 STEC was isolated using the PCR-based culture method (Cho et al., 2006; Dong et al., 2014b). After PCR screening of plates possessing Shiga toxin genes, colonies with these genes were tested for serotype using commercial antiserum (Joongkyeom, Gyeonggi-do, Korea).
For C. jejuni isolation, the standard culture method was used (Dong et al., 2014a). Fecal material (1 g) was homogenized with 9 mL of Bolton broth (Oxoid) supplemented with antibiotics (Oxoid) and 5% laked horse blood and incubated at 42°C for 24 h microaerobically. The enriched broth was then subcultured onto modified charcoal-cefoperazone-deoxycholate agar supplemented with antibiotics (Oxoid). After incubation at 42°C for 48 h, presumptive colonies were transferred onto blood agar for PCR confirmation using a Campylobacter-specific primer (16S rRNA) and a C. jejuni-specific primer (cj0414) (Yamazaki-Matsune et al., 2007).
Sample selection for microbial community analysis
The farm was visited up to seven times during the period of the study. At each visit, 18–34 samples were randomly selected for fecal sampling, and 194 fecal samples were collected. The average fecal collection per cattle was 2.43 ± 1.47 times (ranging from one to six times, median = 2). Among 194 fecal samples, 31 (16.0%) STEC isolates and 47 (24.2%) C. jejuni isolates were identified. Representative samples for microbial community analysis were first selected from the cattle that shed STEC and/or C. jejuni more than twice during the sampling period. The remaining samples were randomly selected from two groups of cattle: STEC shedders and C. jejuni shedders. For the selection of fecal samples from nonshedders, the feces from the same individuals chosen for STEC and/or C. jejuni shedders were first selected, if present, and the remaining cattle were randomly selected from the nonshedding cattle group. Finally, samples were selected from 7 non-shedders, 8 STEC shedders, 8 C. jejuni shedders, and 1 STEC and C. jejuni shedder (total, 24 representative samples).
Pyrosequencing and data analysis
Metagenomic DNA was extracted from 24 representative cows using the FastDNA SPIN Extraction Kit (MP Biomedicals, Santa Ana, CA). The V1–V3 regions of 16S rRNA were amplified by PCR using a bar-coded fusion primer (
Raw sequence data were filtered. Low-quality reads (<25 or >300 bp) were removed and the primer sequences trimmed. Chimera sequences were excluded using the UCHUMI algorithm. Alpha (α) and beta (β) diversity analyses were performed using the CLcommunity software (ChunLab, Inc.). The reads were taxonomically assigned using the EzTaxon-e database (
To evaluate the microbial diversity among 24 fecal samples, an unweighted-pair group method with arithmetic mean analysis (UPGMA) dendrogram was generated based on hierarchical clustering of the Fast UniFrac distance matrix.
Statistical analysis
Differences in the α-diversity and relative abundance of each taxon were investigated between STEC- or C. jejuni-positive and negative, STEC-positive (n = 9) vs. STEC-negative (n = 15), and C. jejuni-positive (n = 9) vs. C. jejuni-negative (n = 15) cattle. The differences were analyzed using the two-tailed Mann–Whitney U test by SPSS statistics, version 22.0 (SPSS IBM, New York, NY). Differences were considered significant at p < 0.05.
Results
α-Diversity of microbial communities
A total of 153,572 reads were obtained from the 24 dairy cattle (Supplementary Table S1; Supplementary Data are available online at
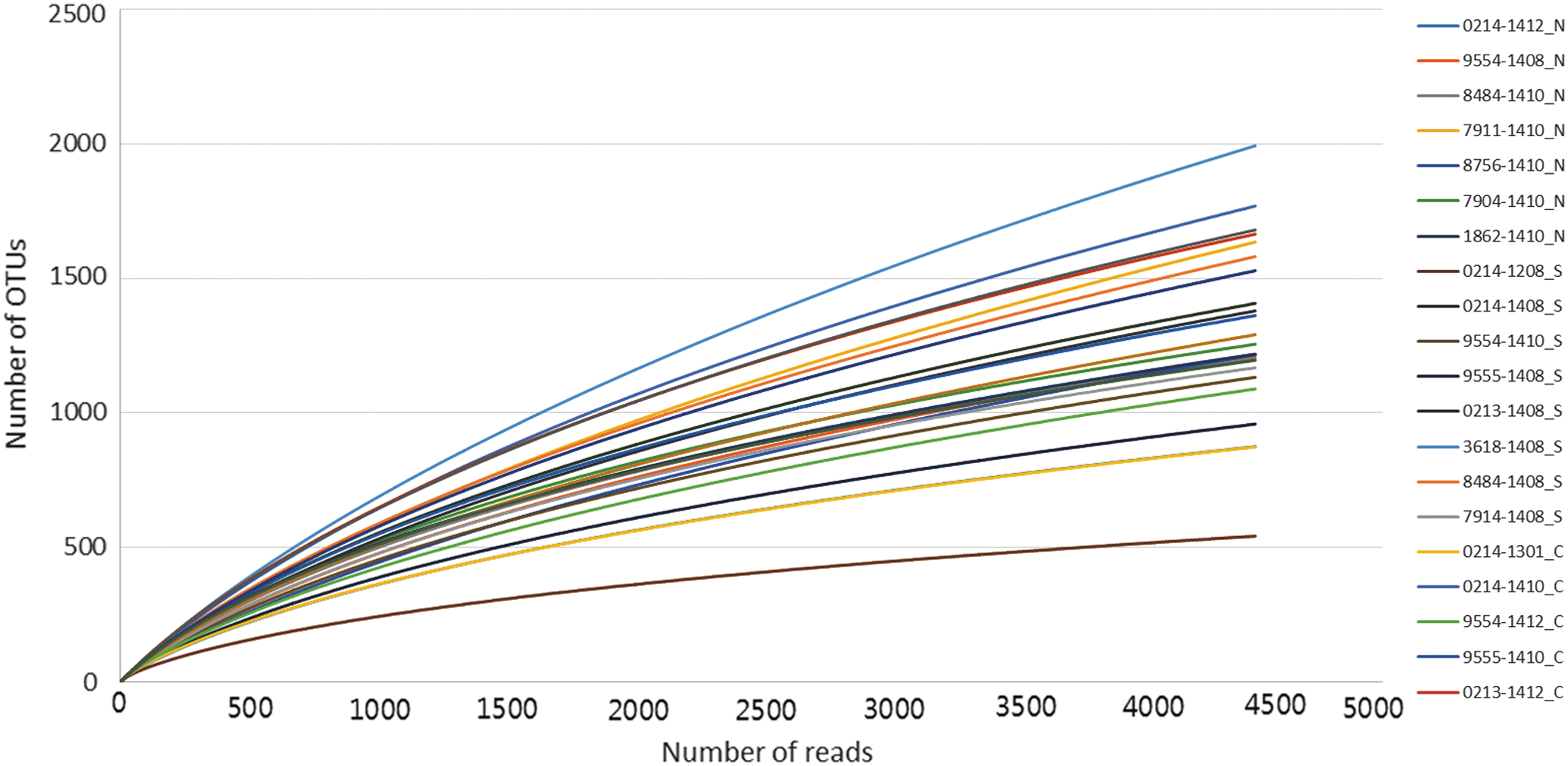
Rarefaction curves for fecal microbial communities of 24 dairy cattle. The curves were truncated after 4390 reads (the minimum across all samples). Color images available online at
α-Diversity was calculated using two different calculation methods, CD-HIT and TBC.
One cow shed both STEC and C. jejuni and was counted in both the STEC-positive and C. jejuni-positive groups.
CD-HIT, cluster database at high identity with tolerance; NP, non-parametric; OTUs, operational taxonomic units; STEC, Shiga toxin–producing Escherichia coli; TBC, taxonomy-based clustering.
Core microbiota
We identified 16 phyla, 33 classes, 64 orders, 151 families, 547 genera, and 1709 species, of which 9 phyla, 13 classes, 18 orders, 47 families, 148 genera, and 261 species were found to constitute the core microbiota in 24 dairy cattle. The core fecal microbiota and the relative abundances in 24 dairy cattle are presented in Supplementary Table S2. A box plot showing the relative abundance of the core microbiota (phyla and genera) in feces from 24 dairy cattle is depicted in Figure 2.
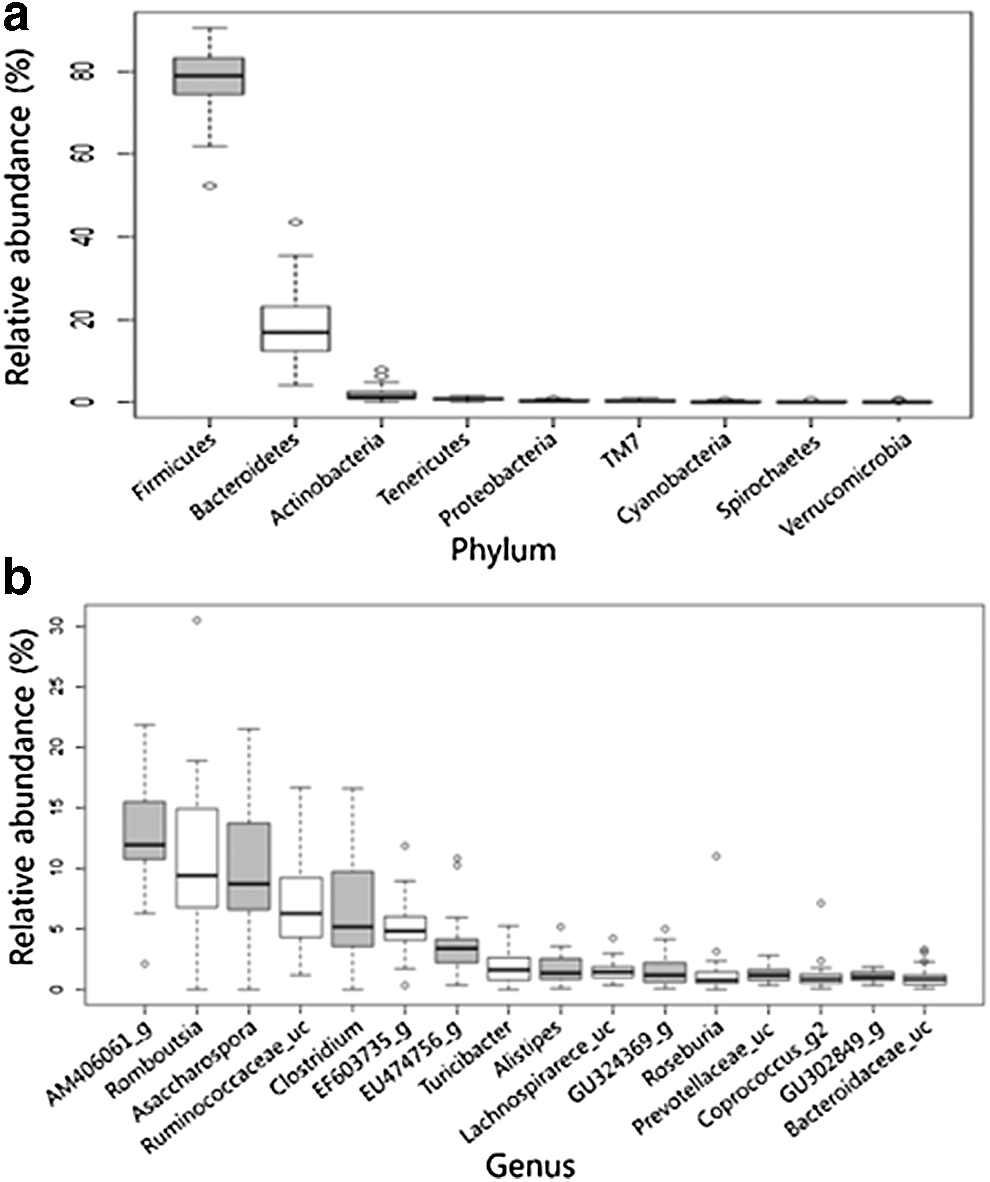
Box plot showing the relative abundance of the core microbiota in feces from 24 dairy cattle.
Fecal bacterial communities of STEC-shedding cattle
In STEC-positive cattle, no significant differences were observed at phylum, class, or order level compared to the STEC-negative cattle. However, at the genus level, the relative abundances of EU474837_g (p = 0.015) and EU475520_g (p = 0.048) were higher, and those of family Clostridiales_us (p = 0.035) and genera Clostridiales_uc_g (p = 0.035), AB506319_g (p = 0.018), and DQ057459_g (p = 0.048) were lower in STEC-positive cattle than in STEC-negative cattle. A higher relative abundance of Ruminococcus (p = 0.064) was observed in the STEC-positive group, but this was not significant (Fig. 3a).
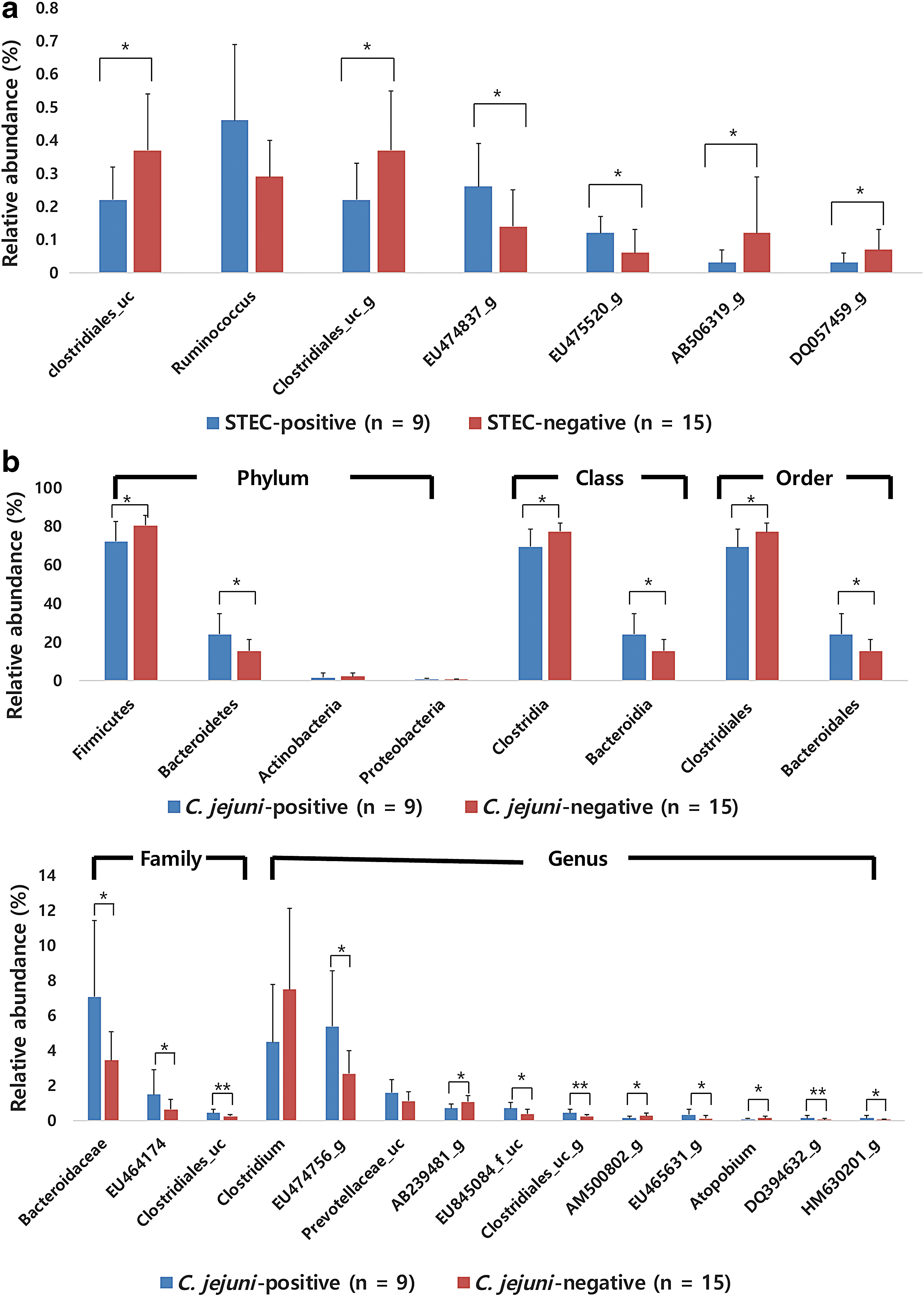
Differences in relative abundances of taxa between cattle positive and negative for foodborne pathogens.
Fecal bacterial communities of C. jejuni-shedding cattle
C. jejuni-positive samples had a significantly lower relative abundance of Firmicutes (p = 0.035) and higher abundance of Bacteroidetes (p = 0.035) than C. jejuni-negative samples. C. jejuni-positive samples had a low abundance of Actinobacteria and high abundance of Proteobacteria, but this was not significant. The class Bacteroidia (p = 0.035); order Bacteroidales (p = 0.035); and families Bacteroidaceae (p = 0.018), EU464174_f (p = 0.021), and Clostridiales_uc (p = 0.01) were more abundant in C. jejuni-positive cattle, whereas the class Clostridia (p = 0.025) and order Clostridiales (p = 0.025) were more abundant in C. jejuni-negative cattle. At the genus level, EU474756_g (p = 0.021), EU845084_f_uc (p = 0.021), Clostridiales_uc_g (p = 0.010), EU465631_g (p = 0.041), DQ394632_g (p = 0.010), and HM630201_g (p = 0.035) were significantly higher in C. jejuni-positive cattle, while AB239481_g (p = 0.012), AM500802_g (p = 0.030), and Atopobium (p = 0.030) were significantly lower (Fig. 3b).
Phylogenetic analysis
Four clusters were generated with the UPGMA method based on 97.5% similarity. While most of the samples belonged to cluster 1, single samples belonged to the remaining clusters: 0214_1208_S, in cluster 2; 1862_1410_C, cluster 3; and 0213_1412_C, cluster 4.
Discussion
In this study, fecal microbial communities were investigated from 24 dairy cattle using NGS techniques. The similarity of our Goods’ coverage value (86.7% ± 5.3%) with that of a previous report (86.9% ± 3.3%) (Xu et al., 2014) suggests that metagenomic samples represent the actual bacterial communities of dairy cattle. Furthermore, the number of OTUs generated was 2.6 times higher than that reported in beef cattle (Durso et al., 2010) and 3.2 times higher than that reported in dairy cattle (Mao et al., 2012). The DIs in our study were 6.2 ± 0.6 and 7.0 ± 0.3 using CD-HIT and TBC methods, respectively; no significant differences in DIs were observed between cattle-shedding STEC or C. jejuni. However, there is no consensus on the diversity of bacterial communities in STEC-shedding cattle. One study reported higher DIs in beef cattle with a high shedding of STEC O157 compared to nonshedders (Xu et al., 2014), whereas the DIs were higher in non-STEC shedders in another study (Zhao et al., 2013). In yet another study, a higher DI was found in high shedders than in low shedders (Aluthge et al., 2014), while a different report found no significant differences in DIs with respect to shedding level (Aluthge, 2015). Differences in DIs between C. jejuni-positive and C. jejuni-negative cattle have not been reported, but higher DIs in C. jejuni-positive groups have been found in chickens (Sofka et al., 2015). Similarly, we observed an increasing trend of DIs in C. jejuni-positive cattle.
Among 16 phyla identified by high-throughput sequencing, 9 were found to be the core microbiota in dairy cattle, regardless of the presence of STEC or C. jejuni. Firmicutes and Bacteroidetes were predominant in all samples, accounting for 96.0% of the bacterial composition. The predominance of these two phyla has been reported in mammalian samples, indicating their vital role in the mammalian gut (Andersson et al., 2008; Durso et al., 2010; O'Donnell et al., 2013). The average relative abundances of Firmicutes and Bacteroidetes were 77.3% and 18.7%, respectively, similar to those of dairy cattle (Ozutsumi et al., 2005), but different from those of beef cattle (Durso et al., 2010; Xu et al., 2014). In beef cattle, a lower concentration of Firmicutes (52.6–62.8%) and higher concentration of Bacteroidetes (29.5–42.1%) were reported (Durso et al., 2010; Xu et al., 2014).
Of 547 genera identified in this study, 148 were found to be the core genera, accounting for more than 90% (90.70–99.10%) of the microbial community. Thus, less than one-third of the taxa account for most of the fecal bacterial communities in dairy cattle. Among the top 10 predominant genera in this study, Clostridium and Alistipes are considered ubiquitous genera in dairy cattle (Dowd et al., 2008), and Peptostreptococcaceae (the family containing Romboutsia and Asaccharospora) and Turicibacter are the core taxa in beef cattle (Shanks et al., 2011), indicating that both beef and dairy cattle might share the same bacterial taxa, but with different relative abundances (Durso et al., 2010).
The microbial impact on STEC shedding is an interesting aspect. Among known genera, Ruminococcus was associated with high-STEC shedding (but not significantly), corresponding to a study comparing high shedders and low shedders of STEC (Fig. 3) (Aluthge et al., 2014). Several genera have also been reported to be associated with STEC shedding in cattle, but the association does not seem consistent. Zhao et al. (2013) suggested that Alistipes might inhibit STEC shedding, while Xu et al. (2014) reported Alistipes to be more abundant in cattle shedding high numbers of STEC. The relative abundance of Prevotella was higher in cattle shedding low numbers of STEC than in high shedders (Aluthge et al., 2014), whereas the opposite was found for high shedders and nonshedders in another study (Xu et al., 2014). In our study, differences in the relative abundance of Alistipes or Prevotella were not observed in cattle regardless of the presence of STEC. This might be ascribed to the use of dairy cattle in our study, because the relative abundance of Prevotella differs between beef and dairy cattle (Durso et al., 2010).
A comparison of intestinal microbial diversity between dairy cattle with and without C. jejuni revealed that the colonization of C. jejuni was not linked to microbial composition (Rapp et al., 2012). However, that study was performed at the herd level rather than for individual cattle. On comparing microbiota between C. jejuni-positive and C. jejuni-negative cattle, the bacterial composition at the phylum level was found to be similar to that of chicken cecal samples with and without Campylobacter (Fig. 3) (Sofka et al., 2015). However, because the comparisons were made between two different species, a bias may exist.
Hierarchical clustering showed that most samples were clustered together, indicating that fecal microbiota of dairy cattle are highly similar. In the clustering, microbial diversity was not related to the shedding of STEC or C. jejuni. This may be because the differences in relative abundances of taxa between pathogen-positive and pathogen-negative individuals were too low to affect microbial diversity. Similarly, minor influences of fecal microbiota on STEC shedding in beef cattle have also been reported (Aluthge, 2015). Microbial diversity might be less affected by the presence of pathogens because the colonization of STEC or C. jejuni in cattle causes asymptomatic infection, and all tested cattle were clinically healthy. Considering that all the samples were collected from the same farm, where animals were managed in a similar manner, the microbial composition might be influenced to a greater extent by management factors rather than individual factors.
Conclusion
In this study, we addressed the fecal microbiota of dairy cattle shedding STEC or C. jejuni. To our knowledge, this is the first study to investigate the fecal microbial diversity in C. jejuni-shedding cattle and STEC-shedding dairy cattle. The core microbiota of dairy cattle revealed that a relatively small number of bacterial species covered most fecal microbiota, indicating their vital role in the intestinal microbial community. In addition, C. jejuni-positive cattle seemed to have more diverse microbiota compared to C. jejuni-negative cattle. Furthermore, several taxa were identified that had a distinguishable relative abundance according to the shedding of foodborne pathogens STEC or C. jejuni, suggesting the possibility to investigate the interaction between pathogens and indigenous bacteria. In-depth investigations of the fecal microbial population would provide essential data for controlling STEC or C. jejuni infection at the farm level. This would have potential applications in the development of strategies to prevent foodborne illnesses in humans.
Footnotes
Acknowledgment
This work was supported by a National Research Foundation of Korea Grant funded by the Korean Government (NRF-2015R1A1A1A05000990).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.