
Abstract
Vibrio parahaemolyticus causes foodborne gastroenteritis, which is often associated with the consumption of raw or undercooked shellfish. Molecular typing can provide critical information for detecting outbreaks and for source attribution. In this study, we describe the development and evaluation of an optimized multiple-locus variable-number tandem-repeat (VNTR) analysis (MLVA) for the characterization of V. parahaemolyticus isolates. The discriminatory power of MLVA was compared to that of pulsed-field gel electrophoresis (PFGE) by typing 73 sporadic isolates. Epidemiologic concordance was evaluated by typing 23 isolates from five epidemiologically well-characterized outbreaks. The optimized MLVA was applied in early warning, epidemiological surveillance, and source tracking for V. parahaemolyticus infections. There was no significant difference in the discriminatory power of PFGE and MLVA with six or eight VNTR loci for the sporadic isolates. All isolates within an outbreak were indistinguishable by MLVA with six loci, except for one outbreak. Typically, the epidemiological survey could be initiated according to PFGE clusters. We applied MLVA with six loci on 22 isolates in two PFGE clusters. Isolates in one PFGE cluster were distinguished by MLVA. Although a follow-up investigation showed that both clusters had no epidemiological concordance, MLVA decreased the frequency of initiation of epidemiological surveys, thereby reducing labor costs. The ability of MLVA to trace the source of infection was evaluated by isolates from two outbreaks and shrimp samples. The isolates from one of outbreaks and a shrimp had the same MLVA type, suggesting that an epidemiological survey was initiated. Data from the epidemiological investigation subsequently indicated that contaminated shrimp from a nearby city (Dongguan) might be the source of the outbreak. In conclusion, these results indicate that the optimized MLVA may be a promising tool for early warning and epidemiological surveillance of V. parahaemolyticus infections.
Introduction
V
At present, molecular typing of the strain is a powerful tool for detecting V. parahaemolyticus infection outbreaks. Molecular typing methods that have been applied to V. parahaemolyticus include ribotyping (Banerjee et al., 2014), direct genome restriction analysis (DGREA) (Ludeke et al., 2014), pulsed-field gel electrophoresis (PFGE) (Chen et al., 2012a), multiple-locus variable-number tandem-repeat (VNTR) analysis (MLVA) (Kimura et al., 2008), multilocus sequence typing (MLST) (Han et al., 2015), clustered regularly interspaced short palindromic repeats (CRISPR) (Sun et al., 2015), and whole-genome sequencing (WGS) (Haendiges et al., 2015). Among them, PFGE is considered the gold standard for subtyping V. parahaemolyticus (Caburlotto et al., 2011) and has been used successfully to identify many foodborne disease outbreaks in the PulseNet database. However, PGFE is labor intensive, time-consuming, and requires a high level of technical expertise, and it is not suitable for typing large numbers of isolates. Recently, Haendiges et al. (2015) utilized WGS to explore the genetic diversity and relationships among V. parahaemolyticus isolates that caused outbreaks in Maryland. WGS provides superior discriminatory power and accurate phylogenetic inferences with high epidemiological correlation; however, the cost and requirement for detailed bioinformatics analysis limit its application in local laboratories.
MLVA is a rapid and highly reproducible typing method that is based on the copy numbers of tandem-repeat units of VNTR loci and can be easily standardized between different laboratories. MLVA has good discriminatory power and high throughput because it is a PCR- and electrophoresis-based technique. Therefore, MLVA is suitable for epidemiological studies of a large number of isolates. MLVA has been applied to the molecular typing of V. parahaemolyticus. A study by Kimura et al. (2008) showed that clonal pandemic O3:K6 strains were distinguishable by MLVA with eight VNTR loci. Subsequently, Harth-Chu et al. (2009) developed an optimized MLVA method based on multiplex PCR of 10 VNTR loci and capillary electrophoresis. Recently, an MLVA assay adapted to high-resolution melt (HRM) analysis was performed within 58 V. parahaemolyticus isolates. HRM-MLVA targeting 12 genes through three separate multiplex real-time PCR reactions was capable of differentiating isolates in the same PFGE cluster and having the same ST (Lüdeke et al., 2015). The reports mentioned above used a range of different loci and various isolates; however, it is not clear whether these loci are really effective in further investigations and whether the loci that clearly differentiate isolates abroad are suited to domestic isolates. The sensitivity and specificity of the VNTR loci should be further evaluated and standardized. In this study, we have developed an MLVA typing scheme that is useful for detecting outbreaks and is easier to perform for typing a large number of isolates.
Materials and Methods
Bacterial isolates
In 2007, a laboratory-based sentinel surveillance of diarrheal infectious diseases was established by the Shenzhen CDC to determine the etiology of acute bacterial diarrheal infection and detect associated outbreaks. One hundred twenty-seven V. parahaemolyticus isolates of various origins, serotypes, isolation year, and PFGE patterns were selected from the surveillance program to develop an MLVA typing scheme. Among these isolates, 73 sporadic isolates were used to analyze VNTR loci and compare the discriminatory power of MLVA and PFGE. Epidemiologic concordance was evaluated by typing 23 isolates from five epidemiologically well-characterized outbreaks. Twenty-two isolates within two PFGE clusters were analyzed for the ability of optimized MLVA to provide early warning of a V. parahaemolyticus outbreak (Supplementary Table S1; Supplementary Data are available online at
Pulsed-field gel electrophoresis
V. parahaemolyticus isolates were subtyped by PFGE as previously described (Parsons et al., 2007). V. parahaemolyticus strains and Salmonella standard strains H9812 were embedded in agarose gel and digested using NotI and XbaI (New England Biolabs), respectively, at 37°C for 4 h. Electrophoresis was performed for 19 h at 6 V/cm with a 10- to 35-s linear ramp time in a Bio-Rad electrophoresis system. A dendrogram was created with BioNumerics 5.1 (Applied Maths) by using the Dice coefficient and the unweighted pair-group method using average linkages (UPGMA), with a 1.5% tolerance limit and 1.5% optimization.
VNTRs PCR amplification
VNTR loci used in this analysis have been described previously by Harth-Chu et al. (2009). However, the primer for the VPTR1 and VP2-07 loci was redesigned. Primers for eight VNTR loci were synthesized and optimized by Invitrogen Co., Ltd. (Table 1). VNTRs were amplified in a 50 μL reaction system containing 0.2 μM of each primer (0.4 μM of each VPTR1 primer) and 25 μL of Go Taq® Green Master Mix. The reaction systems were used according to the different loci. System A: temperature was raised to 95°C for 15 min, followed by 20 cycles of 94°C for 30 s, 62°C for 90 s (per cycle reduces 0.2°C), and 72°C for 60 s, and then followed by 20 cycles of 94°C for 30 s, 58°C for 90 s, and 72°C for 60 s. The final hold was for 30 min at 60°C; System B: temperature was raised to 95°C for 15 min, followed by 30 cycles of 94°C for 30 s, 61°C for 90 s, and 72°C for 60 s. The final hold was for 30 min at 60°C; System C: temperature was raised to 95°C for 15 min, followed by 35 cycles of 94°C for 30 s, 60°C for 90 s, and 72°C for 60 s. The final hold was for 30 min at 60°C. The PCR product was sequenced by Invitrogen Ltd. The resulting data were imported into BioNumerics 5.1 software for clustering analysis, using categorical coefficients of zero tolerance and the unweighted pair-group method with arithmetic mean (UPGMA).
Number of different fragment size polymorphisms detected among 73 sporadic V. parahaemolyticus isolates.
HGI, Hunter and Gaston index; MLVA, multiple-locus variable-number tandem-repeat analysis; VNTR, variable number tandem repeat.
Data analysis and statistics
Diversity of each VNTR locus was assessed using Hunter Gaston Diversity index (Chen et al., 2012b). Diversity of MLVA and PFGE was assessed using Simpson's Diversity index (Olsen et al., 2009). A t-test was used to compare indices for MLVA versus PFGE.
Results
VNTR analysis
Eight VNTR loci (VPTR1, VPTR3, VPTR4, VPTR5, VPTR6, VPTR7, VPTR8, and VP2-07) were selected for the evaluation of an MLVA typing system. The variability of the VNTR loci was confirmed by sequencing. All loci were found to have multiple alleles with substantial variability. Polymorphism at each locus was quantified using the Hunter Gaston Diversity index, which ranged from 0.605 for VPTR3 to 0.964 for VP2-07 (Table 1). Among eight VNTR loci, VP2-07 showed the highest discriminatory ability, in accordance with previous reports (Harth-Chu et al., 2009). No amplification was observed for locus VPTR7 in some non-O3:K6 serotype isolates (33%), especially in the isolates from environment.
Comparsion of the discriminatory power of MLVA with PFGE among the sporadic isolates
Considering the high level of polymorphisms in the VP2-07 locus and no amplification of VPTR7 in some isolates, we performed MLVA with or without these two loci. Seventy-three sporadic V. parahaemolyticus isolates were divided into 68 PFGE patterns and 71 MLVA patterns regardless of whether six or eight loci were used. The Simpson's index of PFGE was 98.40%, while the Simpson's index for MLVA using either six or eight loci was 98.52%. There was no statistical difference between the two methods (t tests, p > 0.05). MLVA with six loci had comparable discriminatory power to MLVA with eight loci, suggesting that six loci may be sufficient for the MLVA assay of V. parahaemolyticus.
Epidemiological concordance analysis
To further evaluate the epidemiological concordance of MLVA with either six or eight loci, 23 isolates from five epidemiologically well-characterized outbreaks were analyzed. All isolates were divided into 12 MLVA patterns with eight loci (Fig. 1A) or six MLVA patterns using only six loci (Fig. 1B). Isolates within an outbreak were indistinguishable by the six-loci MLVA, with the exception of three isolates from a single outbreak that had different MLVA types to four other isolates from the same outbreak. The discriminatory power of our eight-loci MLVA for 23 isolates from outbreaks was too high to distinguish outbreaks. We hypothesized that the VP2-07 locus, with highest diversity, may contribute to the higher discriminatory power of eight-loci MLVA.
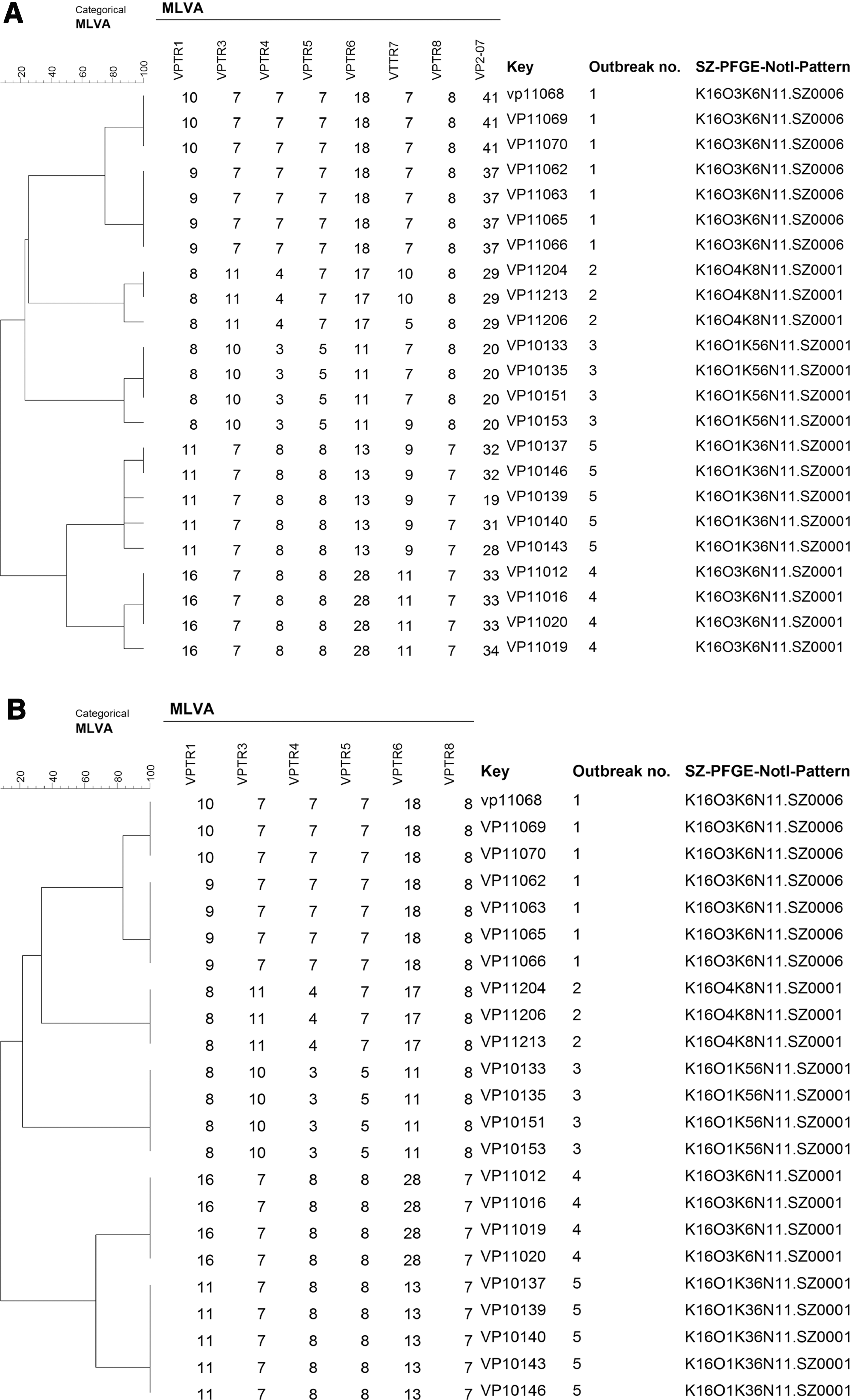
Dendrogram of 23 Vibrio parahaemolyticus outbreak isolates typed by MLVA with eight loci
The combined results of the discriminatory power comparison and epidemiological concordance analysis suggested that MLVA with six loci (VPTR1, VPTR3, VPTR4, VPTR5, VPTR6, and VPTR8) may be more suitable for typing V. parahaemolyticus isolates in local laboratories.
Application of optimized MLVA methods on outbreak detection and source tracking
We applied the six-loci MLVA method to 22 strains from two PFGE clusters (Fig. 2). Based on the result of PFGE, the epidemiological investigation for each cluster should be launched. However, we found that O3:K6 strains with the K16O3K6N11.SZ0001 PFGE pattern had no relationship by MLVA analysis. O3:K6 strains with the K16O3K6N11.SZ0006 PFGE pattern were divided into one main pattern by MLVA, with the exception of three strains. Only an epidemiological survey should be initiated based on the MLVA assay. Although the follow-up investigation showed that both clusters had no epidemiological association, MLVA method can decrease the frequency of surveys, thereby reducing labor costs and effort. This result showed that MLVA had advantages for early warning and initiation of an epidemiological survey for an outbreak, compared to PFGE.
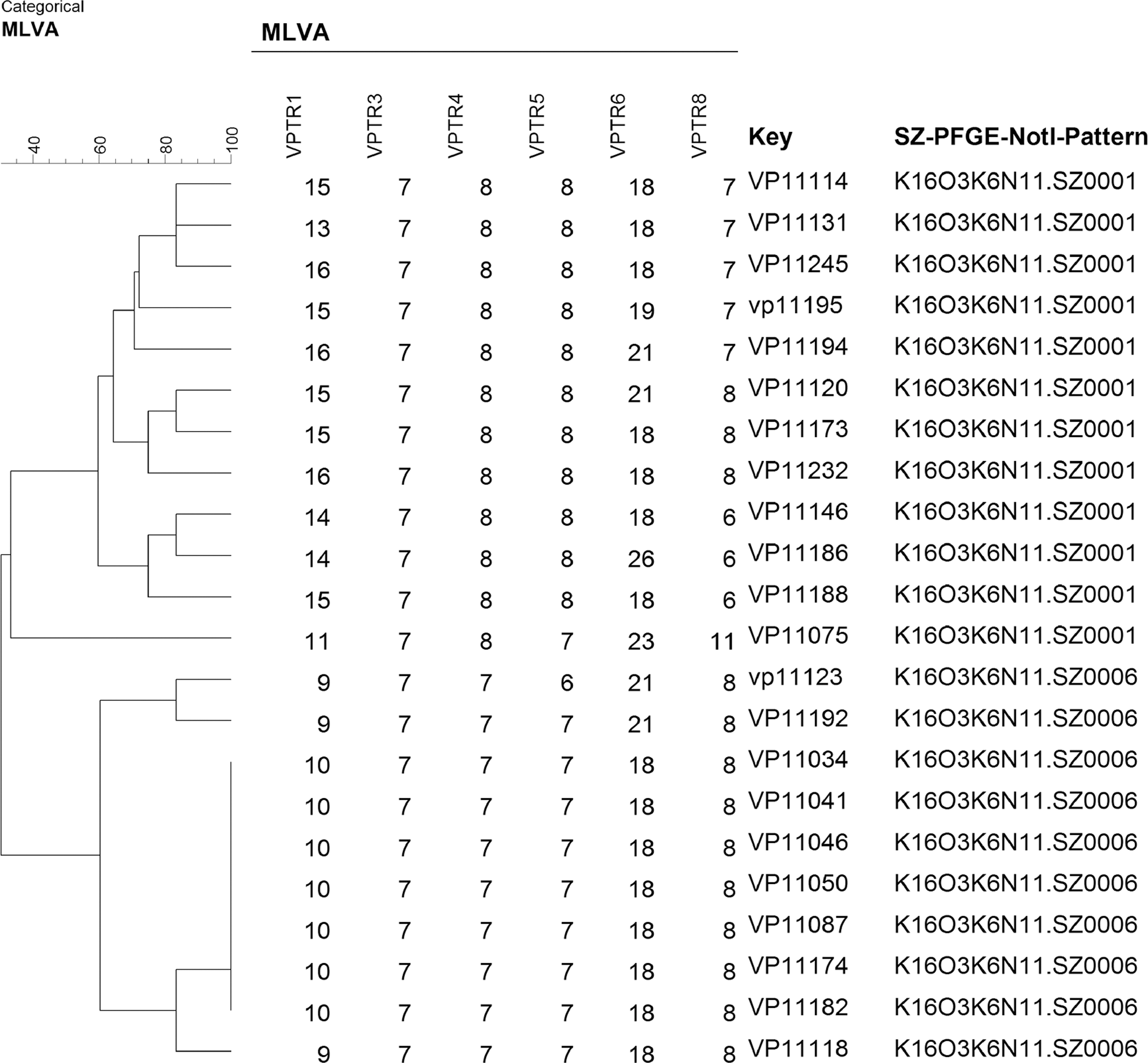
MLVA assay for 22 V. parahaemolyticus isolates within two PFGE clusters. MLVA, multiple-locus variable-number tandem-repeat analysis; PFGE, pulsed-field gel electrophoresis.
We then assessed the ability of MLVA to trace the source of V. parahaemolyticus foodborne outbreaks. In June 2014, two foodborne outbreaks from different districts were reported. We isolated and identified eight V. parahaemolyticus O3:K6 strains from patients' stool samples from two outbreaks (three and five strains, respectively). Meanwhile, a V. parahaemolyticus O3:K6 strain was also isolated from shrimp from the food pollution surveillance system in Shenzhen. PFGE showed that all isolates were of a common type. However, the isolates were divided into two groups by our six-loci MLVA (Fig. 3). Isolates from one of the outbreaks had the same MLVA type as that from the shrimp; therefore, an epidemiological survey was initiated. The results showed that the list of food for the patient in this outbreak contained shrimp, which had the same place of origin (Dongguan) as the shrimp from the food pollution surveillance system. Data from the epidemiological investigation indicated that contaminated shrimp from a nearby city (Dongguan) might be the source of one outbreak and it was necessary to strengthen food safety surveillance in the adjacent district.

MLVA assay for source tracking of V. parahaemolyticus outbreaks. MLVA, multiple-locus variable-number tandem-repeat analysis.
Discussion
Currently, foodborne diseases are one of the top issues affecting public health (Greig and Ravel, 2009; Gould et al., 2013). In 1996, the PulseNet network was built to provide early detection and control of foodborne disease outbreaks in the United States. PulseNet has played an important role in the identification of food pollution sources, tracing the transmission route, and determining prevalence (Lozano-Leon et al., 2003; Li et al., 2012). Detection of foodborne outbreaks relies on a suitable method for subtype determination. The method of choice for the PulseNet surveillance system had been PFGE, although it was time-consuming and labor intensive.
MLVA is a second-generation molecular typing technique, which is widely employed to subtype a variety of species of bacteria (Chiou, 2010; Liu et al., 2016). Several MLVA typing schemes for the characterization of V. parahaemolyticus isolates have been described (Parsons et al., 2007; Harth-Chu et al., 2009). Schemes consisting of eight genomic VNTR loci and 10 genomic VNTR loci have been applied in MLVA for V. parahaemolyticus. Data from Kimura's group have shown that MLVA with eight loci was useful for characterizing highly homogeneous pandemic strains of V. parahaemolyticus. However, their study consisted only of clinical strains of the O3:K6 serotype. In this study, we assessed the MLVA method using a range of clinical and environmental V. parahaemolyticus isolates containing common serotypes from Shenzhen city. In our initial research, we detected 10 VNTR loci previously reported by Harth-Chu et al. Interestingly, two loci with long repeated segments, VP1-10 and VP1-11, could not be amplified from our V. parahaemolyticus isolates. We concluded that the geographic origin of the isolates may have contributed to it. Therefore, these loci were excluded from our MLVA scheme. During VNTR analysis, we found that locus VP2-07 had the highest diversity index (0.964) and showed the greatest discriminatory power between similar isolates. However, the resolution of the VP2-07 locus was too high to cluster the isolates from an outbreak. VPTR7 locus had low amplification efficiency for non-O3:K6 serotype, suggesting that this locus may be more suitable for O3:K6 strains taken from a limited geographical area, such as Shenzhen. Considering the high diversity index of VP2-07 locus and lack of amplification of the VPTR7 locus, the discriminatory power and sensitivity of MLVA were assessed with and without these two loci.
In our analysis, the discriminatory ability of optimized MLVA was studied. For 73 unrelated isolates, MLVA with eight loci or six loci showed similar discriminatory power as PFGE typing and the discriminatory power of MLVA with eight loci or six loci was the same. While measures of discriminatory power are useful in evaluating subtyping methods, epidemiological concordance is equally important for the evaluation of the utility of a typing method. For V. parahaemolyticus outbreak isolates, MLVA typing with six loci clustered each outbreak isolate, except for one outbreak, while MLVA typing with eight loci could not cluster the isolates. The epidemiological consistency analysis indicated that MLVA with six loci was more suitable for the detection of V. parahaemolyticus outbreaks. Data from the sporadic isolates and the outbreaks suggested that we can use fewer VNTRs for an MLVA assay and establish a rapid, easy-to-perform, cost-effective typing method for V. parahaemolyticus.
After removal of the VP2-07 and VPTR7 loci, we applied MLVA typing to isolates from two PFGE clusters. The isolates from a single cluster showed no correlation in the MLVA assay. PFGE is labor intensive and time-consuming, while MLVA is a rapid method that can be easily standardized between laboratories. Therefore, our optimized MLVA could reduce the frequency of initiating epidemiology surveys and provide effective early warning for outbreaks. This result further confirmed Spurgiesz's finding that MLVA was suitable for V. parahaemolyticus outbreak analysis (Spurgiesz et al., 2003). During the subsequent assay, isolates from an outbreak and shrimp were analyzed. Based on the same MLVA type, an epidemiological survey was initiated. Results of epidemiological survey showed that the suspected food in one outbreak contained shrimps, which had the same place of origin as the shrimp collected from food pollution surveillance system. The findings of the survey support the view that the shrimp from the nearby city (Dongguan) may be responsible for the outbreak. This finding provided evidence to support the strengthening of food safety surveillance in the cities surrounding Shenzhen. Overall, these data indicated that MLVA method with six loci was useful for early warning and source tracing of V. parahaemolyticus foodborne disease.
The six-loci MLVA protocol we developed in this study was simple to perform. This was due to the robustness of the technique in terms of simplicity, speed, and reproducibility as opposed to PFGE. Although all V. parahaemolyticus isolates evaluated were from Shenzhen city, the PFGE types of the isolates were representative of the most common PFGE types in Pulsenet China. Nevertheless, a limitation of our study was both the number of outbreak cases and their geographical origin. Although MLVA was able to reduce the frequency of epidemiological survey initiation, there was no epidemiological evidence for the suspected outbreak from the results of the optimized MLVA. Therefore, further studies should widen the scope of this method on outbreak detection and source tracing. An analysis of isolates from different sources and geographical regions is necessary to evaluate MLVA as a global method for V. parahaemolyticus typing.
In conclusion, six loci (VPTR1, VPTR3, VPTR4, VPTR5, VPTR6, and VPTR8) were sufficient to genotypically discriminate sporadic strains and appear suitable for tracing the source of V. parahaemolyticus outbreaks. The optimized MLVA assay developed in this study had comparable discriminatory power to PFGE and high throughput, and may be a viable alternative to PFGE for the subtyping of V. parahaemolyticus isolates.
Footnotes
Acknowledgments
This work was supported by China National Science and Technology Major Projects Foundation (2012ZX10004215-003-005) and National Natural Science Foundation of China (No. 81071433).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.