
Abstract
Capillary electrophoresis–single strand conformation polymorphism (CE-SSCP) coupled with stuffer-free multiplex ligation-dependent probe amplification (MLPA) was developed to identify 13 species of foodborne pathogens simultaneously. Species-specific MLPA probes were designed for nine of these species. These probes were targeted to the groEL, glyA, MMS, tuf, inv, ipaH, nuc, vvh, and 16S rRNA genes, which corresponded to Bacillus cereus, Campylobacter coli, Cronobacter sakazakii, Enterococcus spp., Salmonella spp., Shigella spp., Staphylococcus aureus, Vibrio vulnificus, and Yersinia enterocolitica, respectively. MLPA probes that had been previously developed by our laboratory were used for the other four species (Campylobacter jejuni, Clostridium perfringens, Escherichia coli O157:H7, and Listeria monocytogenes). The CE-SSCP method was optimized to identify all 13 foodborne microbes simultaneously in a single electrogram, in which 50–500 pg genomic DNA was detected per microbe. Twelve species were detected from animal-derived food samples (specifically, milk and sliced ham) that had been artificially inoculated with 12 of the foodborne pathogens, excluding V. vulnificus, which is not usually associated with animal foods. The method developed here could be used as an early warning system for outbreaks of foodborne diseases associated with animal-derived foods in the food industry.
Introduction
F
Although conventional culture techniques are considered to be highly reliable and accurate, they are time-consuming and labor intensive because of the necessity to separately cultivate each target species. Thus, novel technology must be developed to detect potentially harmful bacteria. Polymerase chain reaction (PCR)-based molecular methods for detecting microbes are currently considered to be the most appropriate alternative to conventional methods because they allow comparatively rapid and precise identification.
Various molecular detection methods have been intensively developed over the last decade, evolving from single to multiple detection systems and from qualitative to quantitative types of analyses. Real-time PCR has become popular because it detects multiple pathogens with quantitative data (Mackay, 2004). However, only a limited number of pathogens may be simultaneously detected because only a limited number of fluorescent dyes are available for labeling real-time PCR probes (Klein, 2002).
Multiplex ligation-dependent probe amplification (MLPA) might represent an alternative method. It is a sensitive and reliable technique that simultaneously detects small copy number changes in DNA and RNA sequences (Schouten et al., 2002) and has already been used to detect multiple pathogenic microorganisms (Eijk-Van Os and Schouten, 2011). Detection by MLPA is precise because the two MLPA probes may be ligated where two target-specific probes are precisely hybridized at a target site. Because conventional MLPA is based on the DNA length-dependent discrimination of ligation products, the MLPA probe contains stuffer sequences that generate length differences (Schouten et al., 2002). Consequently, long stuffer sequences cause internal folding and nonspecific binding between the target DNA sequences and probes (Chung et al., 2012a; Shin et al., 2012).
Capillary electrophoresis–single strand conformation polymorphism (CE-SSCP) analysis distinguishes pathogens based on the conformational difference of single-stranded DNA molecules (Andersen et al., 2003). Stuffer sequences in MLPA probes may be excluded when MLPA is combined with CE-SSCP because species-specific sequence-based separation is provided.
This study aimed to develop CE-SSCP coupled with stuffer-free MLPA to target species-specific genetic markers for the detection of 13 foodborne pathogenic bacteria. In addition, the performance of the diagnostic system with artificially contaminated animal-derived foods was evaluated.
Materials and Methods
Bacterial strains and growth conditions
The bacterial strains used in this study are presented in Table 1. Bacillus cereus, Cronobacter sakazakii, Enterococcus spp., E. coli O157:H7, Listeria monocytogenes, Salmonella spp., Shigella spp., Staphylococcus aureus, Vibrio vulnificus, and Yersinia enterocolitica were grown in tryptic soy broth (Difco Laboratories, Detroit, MI) at 37°C for 48 h. Campylobacter coli and Campylobacter jejuni were grown microaerobically in Bolton broth (MB Cell, Los Angeles, CA) at 42°C for 48 h. Clostridium perfringens was grown anaerobically in Hunt broth (MB Cell) at 37°C for 24 h.
Culture Collection of Antimicrobial Resistance Microbes (Seoul, Korea).
Korean Culture Center of Microorganisms (Seoul, Korea).
Nario Agata, Nagoya City Public Health Research Institute (Nagoya, Japan).
American Type Culture Collection (Rockville, MD).
Korean Collection for Type Cultures (Daejeon, Korea).
Sample preparation and DNA isolation
Sterilized milk and ham were purchased at a local market and stored in a refrigerator (4°C) until use, which was within 4 h. All 13 target pathogens were grown separately and mixed with equivalent volumes to obtain 107 colony-forming unit (CFU)/mL of each species. The bacterial strain mixtures were serially diluted in volumes ranging from 102 from 106 CFU/mL and were artificially inoculated onto 10 g of food samples. The samples (10 g) were mixed with 90 mL sterile Buffered Peptone Water (CM0509B; Oxoid, Hampshire, England) in a sterile Stomacher bag. One-milliliter aliquots of the cultivated bacterial suspensions from the broth and food samples were centrifuged at 7500 × g for 10 min. DNA was extracted from the resulting pellets using a DNeasy® Blood and Tissue kit (Qiagen, Valencia, CA) following the manufacturer's instructions. The concentration and purity of the extracted DNA were determined using a microplate reader (Tecan Infinite M200; TECAN, Salzburg, Austria).
Stuffer-free MLPA probe design
MLPA probe sets were designed following the instructions of MRC-Holland (Amsterdam, The Netherlands;
MLPA reaction
The MLPA reaction was performed using a commercial SALSA® MLPA® kit EK1 (MRC-Holland) following the manufacturer's instructions. All MLPA reaction steps were performed with a Veriti™ 96-Well Thermal Cycler (Applied Biosystems, Foster City, CA). A volume of 5 μL genomic DNA (∼10 ng/μL) was denatured at 98°C for 5 min and cooled at 25°C for 5 min. A volume of 1.5 μL SALSA MLPA buffer (10 mM Tris-HCl [pH 8.5], KCl, EDTA, and PEG-6000) and 1.5 μL probe mixture (each 2 fmol/μL) were added to each sample, followed by denaturation at 95°C for 1 min and hybridization at 60°C for 16 h. The ligation reaction was performed at 54°C for 15 min with the ligation mixture (32 μL), which contained ligase buffer A (NAD, pH 3.5), ligase buffer B (Tris-HCl, nonionic detergents, MgCl2, pH 8.5), ligase-65 (glycerol, 0.5% BRIJ, EDTA, 0.1% DTT, KCl, Tris-HCl, polymerase enzyme, pH 7.5), and sterile water. Subsequently, ligase inactivation was performed at 98°C for 5 min.
PCR reaction
PCR reaction was performed to amplify the MLPA products by using the forward primer (5′-HEX-GGGTTCCCTAAGGGTTGGA-3′) and reverse primer (5′-TCTAGATTGGATCTTGCTGGCAC-3′). The PCR reaction mixture (20 μL) contained 10 mM Tris-HCl (pH 9.0), 40 mM KCl, 1.5 mM MgCl2, 1 U Taq DNA polymerase, 0.25 mM dNTPs, 2 μL of MLPA products, 1 μL forward primer (20 pmol/μL), 0.5 μL reverse primer (10 pmol/μL), and sterile water. Initial denaturation was performed at 95°C for 5 min, followed by 35 cycles of amplification (denaturation at 95°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s), and a final extension at 72°C for 7 min. The MLPA products were electrophoresed on 3% agarose gel with 0.5× Tris-borate-EDTA (TBE) buffer (20 mM Tris-HCl, 10 mM boric acid, and 0.5 mM Na2EDTA, pH 8.0), stained with ethidium bromide (0.5 μg/mL), and then visualized with an ultraviolet transilluminator (Cleaver Scientific Ltd., Warwickshire, England).
CE-SSCP analysis
CE-SSCP analysis was performed with PCR products (1 μL) that contained 8.8 μL deionized formamide (Hi-Di Formamide; Applied Biosystems) and 0.2 μL GeneScan ROX 500 size standard (Applied Biosystems). The samples were denatured at 95°C for 5 min and immediately cooled on ice for 3 min. CE-SSCP was performed with an ABI 3130XL genetic analyzer (Applied Biosystems) using a 50 cm × 50 μm capillary array (Applied Biosystems) and 15% (w/v) solution of Pluronic F108 (Sigma-Aldrich, St. Louis, MO). The polymer matrix was prepared by dissolving 15% (w/v) Pluronic F108 PEO-PPO-PEO triblock copolymers (Sigma-Aldrich) in 0.7% (w/v) EDTA buffer (Applied Biosystems). Electrophoresis was performed under the following conditions: an injection voltage of 15 kV, an electrophoresis voltage of 15 kV, an injection time of 5 s, a running time of 70 min, and a constant temperature of 35°C. The signals were analyzed by DNA analysis software (GeneMapper; Applied Biosystems). The elution time and the area of each peak on the chromatograms were determined by arbitrary units (AU). Each individual peak was identified by the relative migration compared with ROX (red) 500 size standards (Applied Biosystems).
Results
Design of specific stuffer-free MLPA probes
The species-specific sequences of each target strain were employed as markers for the individual detection of each species. Target foodborne pathogens included B. cereus, C. coli, C. jejuni, C. perfringens, C. sakazakii, E. coli O157:H7, L. monocytogenes, Salmonella spp., Shigella spp., S. aureus, V. vulnificus, Enterococcus spp., and Y. enterocolitica. For 9 of the 13 strains (namely B. cereus, C. coli, C. sakazakii, Enterococcus spp., Salmonella spp., Shigella spp., S. aureus, V. vulnificus, and Y. enterocolitica), new MLPA probes were designed to target the specific genes (Table 2). In addition, 4 MLPA probe sets that were designed in a previous study (Chung et al., 2012b) were used because the proper target genes were selected, facilitating simultaneous detection in a single run when mixed with all 13 pathogens. Specific MLPA probes for the 13 target bacterial marker genes were designed, with the product sizes being shown in Table 2. The MLPA products of the pathogens that were amplified by each specific MLPA probe ranged from 93 to 115 bp in size (Table 2).
Amplicon size includes the left hybridization sequence, right hybridization sequence, and both common primers.
Identification and simultaneous detection of foodborne pathogens using MLPA-CE-SSCP
The electropherogram of each foodborne pathogen that was analyzed using MLPA-CE-SSCP is shown in Figure 1. All 13 target microbes were identified from a single peak in relative migration time (Fig. 1) and were also analyzed simultaneously from a single reaction. The peaks of the 13 strains were separated from one another (Fig. 2). The peaks corresponding to each individual strain from the pathogen mixture had identical retention times to those observed when single strains were analyzed. The identification ability of the identical peaks corresponding to each species was verified with MLPA-CE-SSCP by systemically changing the concentrations of each strain. This technique resulted in changes to the peak area corresponding to that target strain, without affecting the other peaks. Even though the absolute migration time of a target slightly changed in each run, the relative migration was reproducibly identical when compared with the ROX 500 size standards.
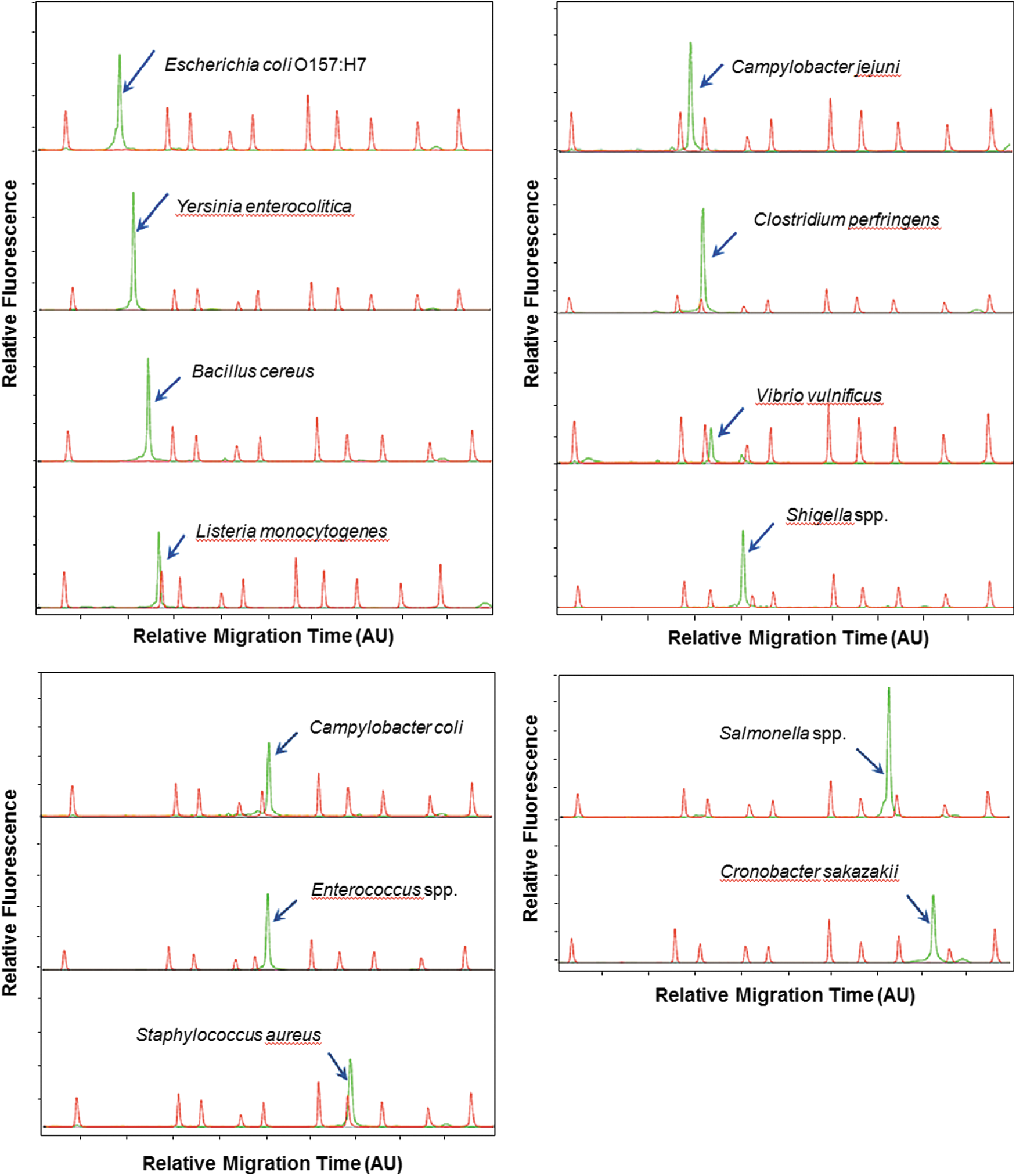
Electropherograms obtained from the CE-SSCP analysis of the MLPA products for each pathogen. Relative migration time (x-axis) and fluorescence intensity (y-axis) are shown as AU and relative fluorescence units, respectively. AU, arbitrary units; CE-SSCP, capillary electrophoresis–single strand conformation polymorphism; MLPA, multiplex ligation-dependent probe amplification.
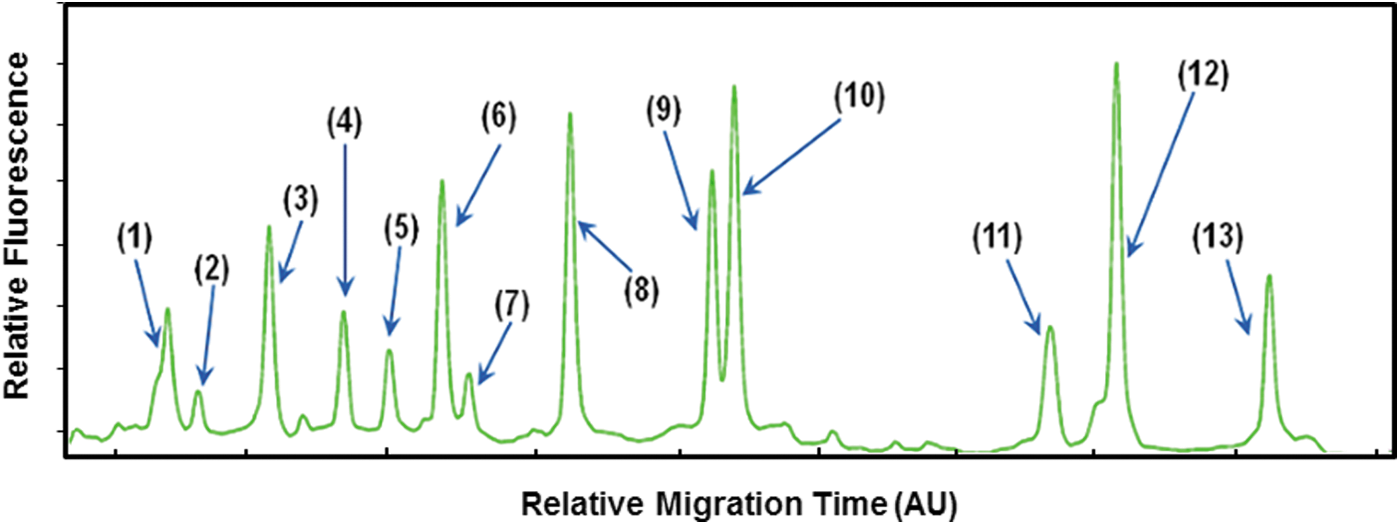
Electropherogram obtained from the CE-SSCP analysis of the multiplex MLPA products for 13 foodborne pathogens. (1) Escherichia coli O157:H7, (2) Yersinia enterocolitica, (3) Bacillus cereus, (4) Listeria monocytogenes, (5) Campylobacter jejuni, (6) Clostridium perfringens, (7) Vibrio vulnificus, (8) Shigella spp., (9) Campylobacter coli, (10) Enterococcus spp., (11) Staphylococcus aureus, (12) Salmonella spp., and (13) Cronobacter sakazakii.
The detection limit was examined by performing MLPA on serially diluted genomic DNA concentrations, ranging from 10 ng to 1 pg, for all pathogens tested in this study (Table 1). Nine microbes (namely E. coli O157:H7, C. jejuni, C. perfringens, L. monocytogenes, S. aureus, Y. enterocolitica, C. sakazakii, Shigella spp., and Salmonella spp.) had a detection limit of 50 pg. The detection limit of the other four microbes (namely B. cereus, C. coli, V. vulnificus, and Enterococcus spp.) was 500 pg, resulting in lower detection efficiency. Because 1–100 pg of genomic DNA per microliter is the equivalent of 101–103 CFU/mL (Zheng et al., 2004), all strains had detection ranges of about 5 × 102–103 CFU/mL, without the pre-enrichment of target samples to reach the detection level of bacteria.
Application of the developed method in a real food system
The developed method was applied to two real foods, milk and ham, to evaluate the detection ability of the method. Because the absolute migration time of each peak was reproducible, infection could be identified using mobility characteristics. The results of various multiple infections are shown in Figure 3. Except for V. vulnificus, the peaks inoculated with the other 12 pathogens were identified from their relative mobility.
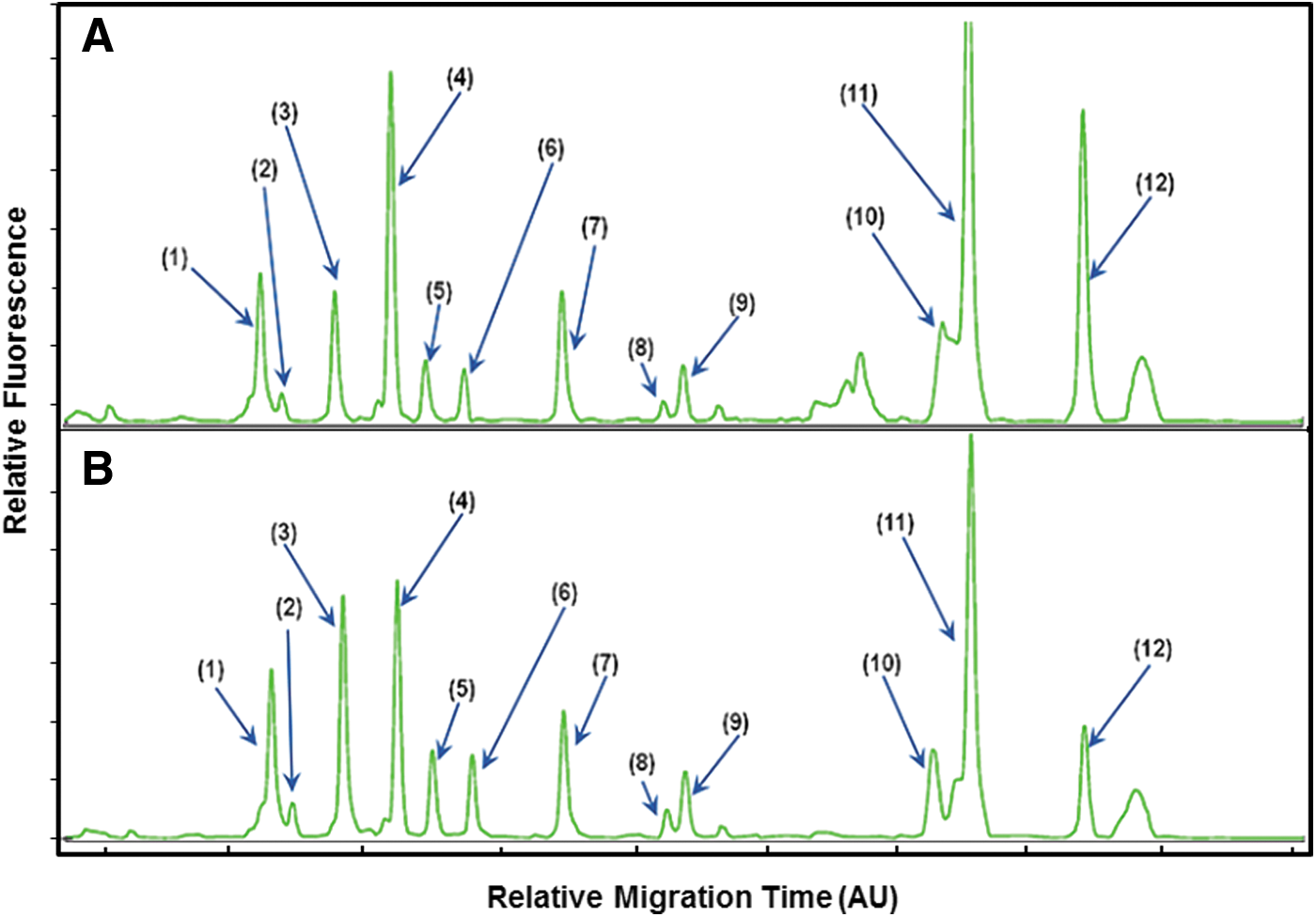
Electropherogram obtained from the CE-SSCP analysis of the MLPA products for 12 foodborne pathogens artificially incubated in milk
Discussion
Conventional culture techniques are still primarily used to identify foodborne pathogens. However, these techniques typically require 3–7 d to complete (Kim et al., 2007), potentially requiring 7–10 d if subcultures are required to confirm identification through biochemical tests (Gehring and Tu, 2011). Conventional techniques are also labor intensive and expensive because of the need to separately cultivate each target species. Recently, PCR-based detection methods were introduced to overcome these drawbacks. Multiplex PCR coupled with various detection methods and real-time PCR has greatly improved the speed of detecting microbes. However, the detection capability of these methods is restricted to a few targets in a single tube because of the low resolution of agarose gels in traditional PCR or limited availability of spectrally distinct fluorescence tags in real-time PCR (Grace et al., 2003; Naravaneni and Jamil, 2005; Yamazaki-Matsune et al., 2007; Suo et al., 2010). To overcome this problem, CE-SSCP coupled with multiplex PCR analysis was developed in our previous study (Kim et al., 2010) because it provides much higher resolution than agarose gel. This analysis takes <5 h to detect seven pathogens in food. However, the efficacy of amplifying and optimizing reaction conditions was difficult because multiple primer sets are required. Furthermore, false-positive results might be obtained when using the 16S rRNA gene as the target gene because it exhibits a high degree of similarity among foodborne pathogens (Kim et al., 2008). Therefore, this study used an MLPA technique with two specific probes. Because ligation only occurs when two probes hybridize precisely and ligated probes are amplified by common primers, nonspecific probe hybridization is minimized (Chung et al., 2012a). Conventional MLPA probes use stuffer sequences to separate length differences. Length differences affect the hybridization efficiency of probes, and may adversely affect the amplification step because short products have a high tolerance of PCR inhibitors, and are amplified with greater specificity compared with long products (Shin et al., 2012). To resolve the problem, we employed CE-SSCP separation, which facilitated DNA sequence-based separation based on differences in the genomic sequences of individual microbes. The resolution of separation by CE-SSCP was also improved by using a PEO-PPO-PEO triblock copolymer rather than a conventional polymer matrix, GeneScan™, when running the CE (Shin et al., 2012). Digital PCR was introduced within the last year as novel technology for rapid and sensitive detection of pathogens. Bian et al. (2015) demonstrated that 10 CFU/mL of E. coli O157:H7 and L. monocytogenes may be detected within 2 h in an artificially contaminated drinking water sample. Even though this method is very sensitive, the cost of consumables and instruments is very expensive, limiting the feasibility of using this technique in many laboratories at present.
This study used a model set of 13 pathogens, adding 4 strains to the 9 strains considered to be major foodborne pathogens. Among the nine major foodborne pathogens, we changed the five MLPA probes corresponding to B. cereus, Salmonella spp., Shigella spp., Y. enterocolitica, and S. aureus from those used in our previous study (Chung et al., 2012b) to newly designed probes for several reasons. Specifically, B. cereus produces an emetic toxin and several diarrheal toxins (Rasko et al., 2005). Because the previously designed MLPA probe for B. cereus only detected the emetic toxin, a new version of the B. cereus MLPA probe was designed to target groEL genes, facilitating the detection of both the emetic and diarrheal strains of B. cereus. The new probe to target inv gene allows Salmonella to be detected to the genus level (Rahn et al., 1992), whereas only Salmonella enterica serotype Enteritidis could be detected by the MLPA probe developed in the previous study. We also targeted the ipaH gene to detect all four serotypes of Shigella (Warren et al., 2006), rather than just detecting Shigella flexneri. For Y. enterocolitica, we decided to use a specific region of the 16S rRNA gene rather than the yst gene because it is possible to detect pathogenic Y. enterocolitica that does not have the yst gene. A new MLPA probe was designed by using different sequences from a previously designed MLPA probe in the nuc gene to separate the peak of S. aureus from that of E. coli O157:H7. The extra four strains included C. sakazakii, C. coil, V. vulnificus, and Enterococcus spp. C. sakazakii is a pathogenic bacterium that causes foodborne diseases (Iversen and Forsythe, 2003). The target gene for the MLPA probe of C. sakazakii is the dnaG gene on the macromolecular synthesis (MMS) operon, which is only found in C. sakazakii (Seo and Brackett, 2005). C. coli cause gastrointestinal infections similar to C. jejuni (Acheson and Allos, 2001). We used the glyA gene for the C. coli MLPA probe because it differentiates C. coli from C. jejuni (Wang et al., 2002). V. vulnificus is pathogenic and causes vomiting, diarrhea, and abdominal pain in infected patients. The V. vulnificus-specific hemolysin gene, vvh, was used as the target gene when designing the MLPA probe for this disease (Jones et al., 2012). Enterococcus spp., including E. faecalis, may infect animal foods through fecal contamination of the animal and sometimes show vancomycin-resistant activity (Giraffa, 2002). The tuf gene was used as the target gene for the MLPA probe because it detects Enterococci at the genus level (Ke et al., 1999).
In conclusion, this study developed a diagnostic system that combined high-resolution CE-SSCP with MLPA using stuffer-free probes to detect 13 foodborne pathogens. Specific laboratories may select several strains of the 13 pathogens that they test for regularly. The system may be used by the food industry as an early warning system for pathogen-associated food poisoning because it identifies target foodborne pathogens with greater speed compared with conventional methods. However, further research is required to improve the sensitivity of the system, separately or in combination with optimized protocols, for the pretreatment of food samples and/or multiplex displacement amplification. Furthermore, the system must be optimized for a variety of foods and for each target pathogen because some food matrices and interactions between foods and pathogens may have a negative impact on the system and, hence, its performance.
Footnotes
Acknowledgments
This work was carried out with the support of the Cooperative Research Program for Agriculture Science and Technology Development (Project No. 00932901) and was supported by the Internship Program of National Institute of Animal Science, Rural Development Administration (Korea).
Disclosure Statement
No competing financial interests exist.