
Abstract
The enterotoxin gene cluster (egc) has been proposed to contribute to the Staphylococcus aureus colonization, which highlights the need to evaluate genetic diversity and virulence gene profiles of the egc-positive population. Here, a total of 43 egc-positive isolates (16.2%) were identified from 266 S. aureus isolates that were obtained from various food and clinical specimens in Shanghai. Seven different egc profiles were found based on the polymerase chain reaction (PCR) result for egc genes. Then, these 43 egc-positive isolates were further typed by multilocus sequence typing, pulsed-field gel electrophoresis (PFGE), multiple-locus variable-number tandem-repeat analysis (MLVA), and accessory gene regulatory (agr) typing. It showed that the 43 egc-positive isolates displayed 17 sequence types, 28 PFGE patterns, 29 MLVA types, and 4 agr types, respectively. Among them, the dominant clonal lineage was CC5-agr II (48.84%). Thirty toxin and 20 adhesion-associated genes were detected by PCR in egc-positive isolates. Notably, invasive toxin genes showed a high prevalence, such as 76.7% for Panton–Valentine leukocidin encoding genes, 27.9% for sec, and 23.3% for tsst-1. Most of the examined adhesion-associated genes were found to be conserved (76.7–100%), whereas the fnbB gene was only found in 8 (18.6%) isolates. In addition, 33 toxin gene profiles and 13 adhesion gene profiles were identified, respectively. Our results imply that isolates belonging to the same clonal lineage harbored similar adhesion gene profiles but diverse toxin gene profiles. Overall, the high prevalence of invasive virulence genes increases the potential risk of egc-positive isolates in S. aureus infection.
Introduction
S
Extensive studies have showed that egc genes are the most prevalent SE genes in both commensal and invasive S. aureus strains (Becker et al., 2004; Nowrouzian et al., 2011). Although all egc-encoding SEs have shown superantigen and emic activity (Jarraud et al., 2001; Omoe et al., 2013), egc genes are rarely found in toxin shock and staphylococcal foodborne illness cases (Ferry et al., 2005). Moreover, antibodies against egc SEs are rarely raised in serum from healthy humans (Holtfreter et al., 2004), and egc-positive strains have shown increased capacity for long-term persistence in the infantile gut (Nowrouzian et al., 2011). Therefore, it seems, in general, that egc probably contributes to the colonization of S. aureus (Ferry et al., 2005). It is well known that S. aureus infections are generally preceded by commensal colonization (Bogaert et al., 2004). Thus, it is essential to evaluate the virulence gene profiles of the egc-positive population.
Traditionally, multilocus sequence typing (MLST) and spa typing have been used to characterize the genetic background of egc-positive isolates (Blaiotta et al., 2006; van Belkum et al., 2006; Collery et al., 2009). The occurrence of the egc locus is strongly associated with clonal lineages (Chini et al., 2006; van Belkum et al., 2006). However, systematic data on the genetic diversity of the egc-positive population are rare. In this study, we used a developed MLVA protocol (Brandt et al., 2013) to explore the genetic diversity of egc-positive isolates obtained from various food and clinical specimens in Shanghai, and we compared the performance of MLVA with that of MLST and pulsed-field gel electrophoresis (PFGE). Furthermore, we studied the polymorphisms of the egc locus, virulence gene profiles of egc-positive isolates, and their association with molecular typing features.
Materials and Methods
S. aureus isolates harboring egc
A total of 266 S. aureus isolates were obtained from various food samples and clinical specimens during a 2-year period (July 2010 to October 2012) in Shanghai. This collection comprised 142 isolates that originated from various food samples, such as milk, meats, vegetables, and fruits, which were randomly purchased from local grocery stores; 124 isolates obtained from clinical specimens, such as blood, pus, and sputum, including 22 isolates obtained from human clinical vomit in staphylococcal foodborne illness cases. Isolation and identification of S. aureus were performed as previously described (Wang et al., 2012).
Genomic DNA of each isolate was purified using TIANamp Bacteria DNA Kit (TIANGEN Biotech) following the manufacturer's instructions, with the addition of lysostaphin (Sigma-Aldrich) for bacterial lysis. DNA amount and purity were tested with an NA-2000 Spectrophotometer (Thermo Fisher Scientific). Then, all isolates were rapidly screened for the presence of the egc locus by using a real-time polymerase chain reaction (PCR) method (Fusco et al., 2011). In parallel, PCR detection of the egc-encoding SE genes (seg, sei, sem, sen, seo, and selu) was also performed, using primers listed in Supplementary Table S1 (Supplementary Data are available online at
Detection of toxin and adhesion genes
The egc-positive isolates were further tested by simplex PCR amplification of the other 24 toxin genes (Supplementary Table S1) and 20 adhesion-associated genes (Supplementary Table S2) using previously described primers. The tested 24 toxin genes included enterotoxin genes (sea, seb, sec, sed, see, seh, sej, sek, sel, sep, seq, ser, ses, and set), toxic shock syndrome toxin genes (tsst-1), hemolysin-encoding genes (hla, hlb, hld, hlg, and hlg v), exfoliative toxin genes (eta, etb, and etd), and Panton–Valentine leukotoxin genes (lukS-PV and lukF-PV). The selected adhesion-associated genes were bbp, clfA, clfB, can, ebps, eno, Fib, fnbA, fnbB, isdA, sasG, sdrC, sdrD, sdrE, icaA, icaD, efb, sak, tagO, and tarK. Furthermore, the type of accessory gene regulatory (agr) system was determined by multiplex PCR (Gilot et al., 2002).
Multilocus sequence typing
All egc-positive isolates were further characterized by MLST. Seven S. aureus housekeeping genes (arcC, aroE, glpF, gmk, pta, tpi, and yqiL) were amplified by PCR, and DNA sequencing was performed for all of the PCR products (Enright et al., 2000). DNA sequence of each target gene was analyzed using the MLST website (
SmaI-PFGE
SmaI-PFGE analysis was performed for all egc-positive isolates according to a standard laboratory protocol developed by the PulseNet for S. aureus (
MLVA
MLVA was performed as previously described by Brandt et al. (2013). Two separate multiplex PCR with different fluorescently labeled primer sets were prepared. After PCR, products were diluted 1:100 in MilliQ water. One microliter of the diluted samples was mixed with 0.2 μL of 1200 LIZ size standard and 10 μL of HIDI formamide (Applied Biosystems). After heat denaturation at 95°C for 5 min, the fragments were separated on an ABI 3500 genetic analyzer system. The results were analyzed in the GeneMaper 4.0 software (Applied Biosystems). If a VNTR locus was not detected during fragment analysis, PCR for that particular locus was repeated in a singleplex reaction. Loci that did not yield a peak were assigned number 99.
Statistical analysis
Statistical analysis was computed using SPSS v.18.0 (SPSS, Inc.). Pearson's chi-square test (two-tailed) was used to analyze the difference of the distribution of virulence genes among egc-positive isolates from foods and clinical specimens. The difference is considered significant if the p-value is less than 0.05.
Results
Identification of egc locus
The result showed that a total of 43 isolates (16.17%) were positive for the egc locus (Table 1). The occurrence of the egc locus among the isolates from clinical specimens (18.55%) was slightly higher than that among the isolates from food (14.08%). Seven different egc profiles were identified among the 43 egc-positive isolates, and the percentages of egc profile1 (seg, sei, sem, sen, and seo) and egc profile 2 (seg, sei, sem, sen, seo, and selu) were 39.53% (17/43) and 48.54% (21/43), respectively. The remaining five egc profiles lacked one or more egc-encoding SE genes (Table 1). Interestingly, all five egc-positive isolates obtained from a patient's vomit in staphylococcal foodborne illness cases carried egc profile1 (Fig. 1, vomit).
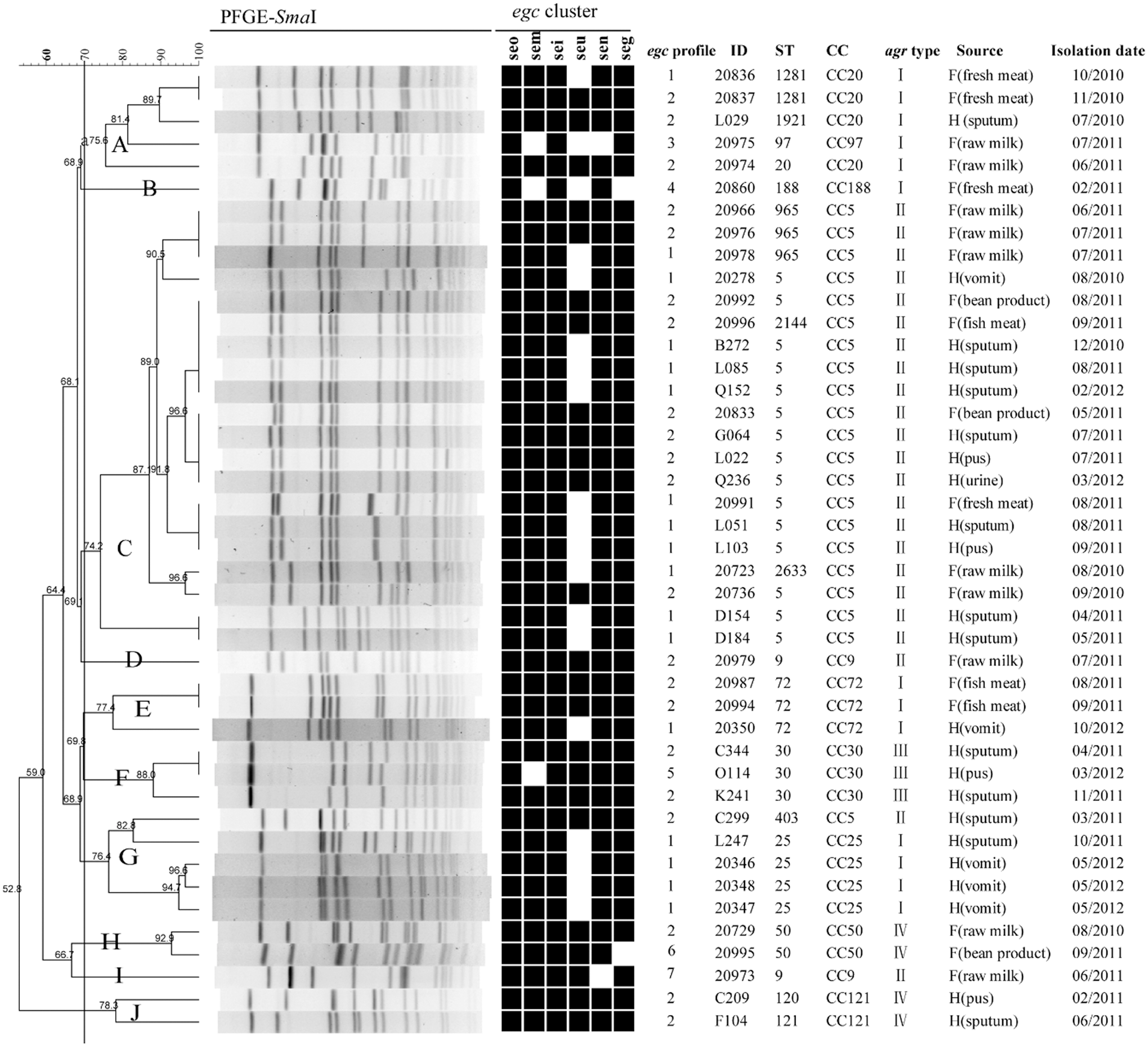
Dendrogram of PFGE patterns and genotypic relationship of 43 Staphylococcus aureus egc-positive isolates. The cluster cutoff was set at 70% similarity. For each isolate, egc profile, MLST, clonal complexes, agr types, source of isolates, and isolation date are listed. H means “Human,” and F means “Food.” Black box, positive; white/blank, negative. MLST, multilocus sequence typing; PFGE, pulsed-field gel electrophoresis.
The clonality of egc-positive isolates
The clonality of 43 egc-positive isolates is shown in Table 2. The result showed that 43 egc-positive isolates were distributed in 17 different STs and 4 agr types. The MLST data analysis by eBURST grouped these isolates into 10 clonal complexes (CCs). Among them, CC5-agr II was the most prevalent (48.84%) clonal lineage. Then, the association of clonality with isolate origin was analyzed. The results showed that CC9-agr II, CC50-agr IV, and CC97-agr I were found in isolates from food. In contrast, ST5-agr II, CC25-agr I, CC30-agr III, and CC121-agr IV were mainly identified in clinical isolates. The egc-positive isolates obtained from a patient's vomit in staphylococcal foodborne illness cases belonged to ST5, ST25, and ST72, respectively.
PFGE of egc-positive isolates
Of these 43 egc-positive isolates, PFGE analysis revealed 28 different PFGE patterns and 10 PFGE clusters that were designated by symbols A to J (Fig. 1). The most predominant PFGE cluster was cluster C, consisting of 20 (46.51%) isolates belonging to CC5. Many isolates belonging to the same clonal complex were grouped together by PFGE patterns. However, there were some exceptions, such as two CC9 isolates, which were grouped into two distinct clusters D and I. As shown in Figure 1, many egc-positive isolates with different egc profiles and origins were grouped into the same PFGE cluster, especially in clusters A and C; therefore, our data demonstrated that there was no significant correlation among PFGE patterns, egc profiles, and isolate origins.
MLVA of egc-positive isolates
Based on MLVA, 43 egc-positive isolates were grouped into 29 MLVA types (Fig. 2). Among them, 4 MLVA types were submitted as new registrations to the MLVA database. The relationship between MLVA and MLST results is shown in Figure 2. The isolates of the same clonal complex were significantly grouped together by MLVA dendrogram. MLVA showed a higher resolution than MLST. For example, 21 isolates of CC5 distributed in 4 STs could be assigned into 14 MLVA types.
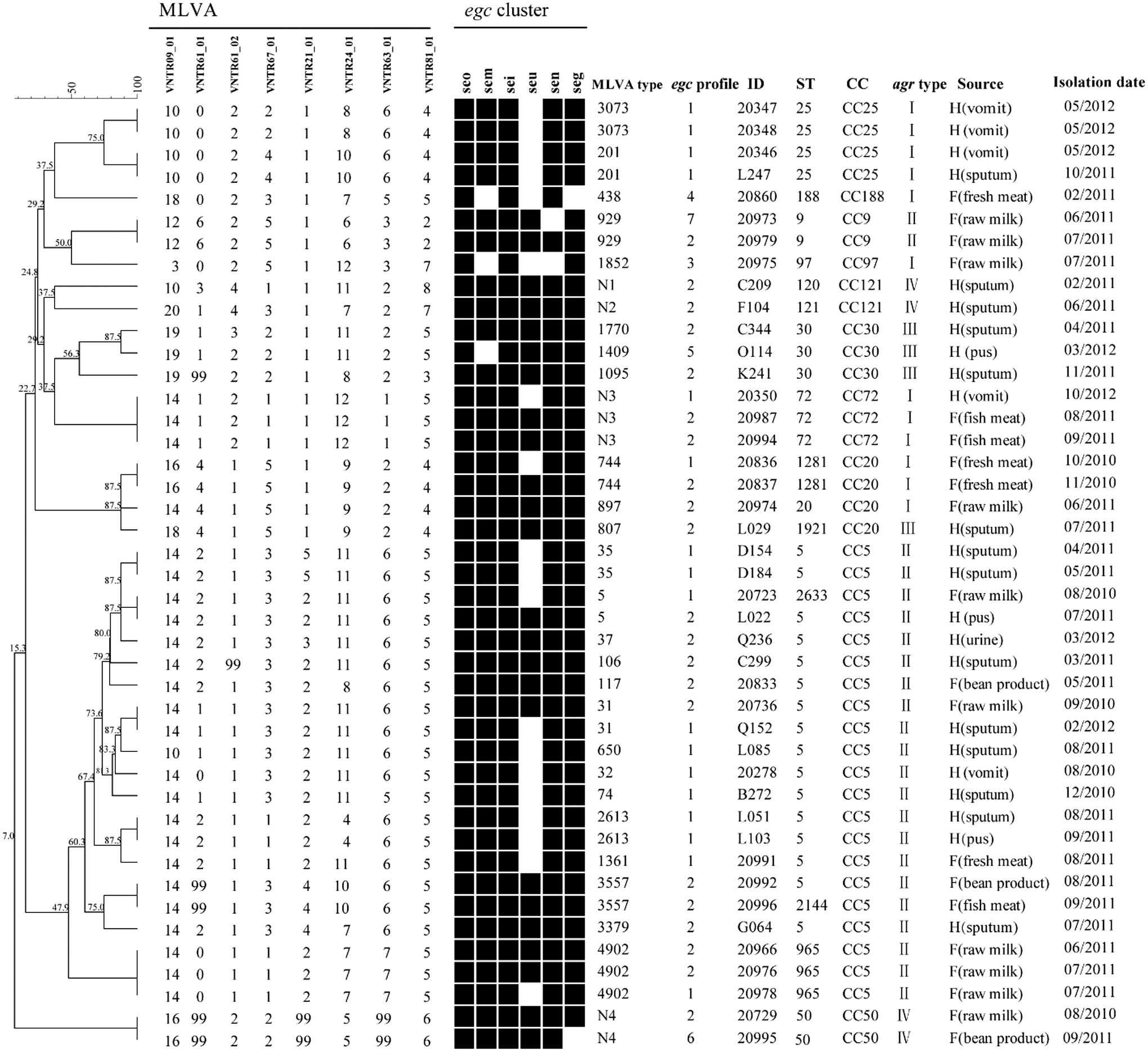
Dendrogram of MLVA types and genotypic relationship of 43 S. aureus egc-positive isolates. For each isolate, egc profile, MLST, clonal complexes, agr types, source of isolates, and isolation date are listed. H means “Human,” and F means “Food.” Black box, positive; white/blank, negative.
Prevalence of toxin and adhesion genes
The presence of 30 toxin genes and 20 adhesion genes in the 43 egc-positive isolates is shown in Figure 3. For toxin genes, the PVL-encoding genes were highly prevalent (76.7%) in egc-positive isolates. TSST-1-encoding gene (tsst-1) was found in 10 (23.3%) clinical isolates. Genes encoding exfoliative toxins were either absent (etb) or rare (eta 2.3%, etd 11.4%) in the examined isolates. The SE gene sec was present in 27.9% of the isolates. Notably, hemolysin genes hla, hlb, hld, and hlg v were found in all tested isolates. Statistical analysis revealed that only tsst-1 gene was more significantly prevalent among clinical isolates compared with the isolates from food. For the tested adhesion-associated genes, the genes clfB, eno, fib, isdA, sdrC, sdrD, icaA, icaD, efb, sak, tagO, and tarK were identified in all egc-positive isolates; the genes bbp, clfA, Cna, ebpS, fnbA, sasG, and sdrE were found in most isolates (76.7–97.7%); and fnbB was only found in 8 egc-positive isolates (18.6%).
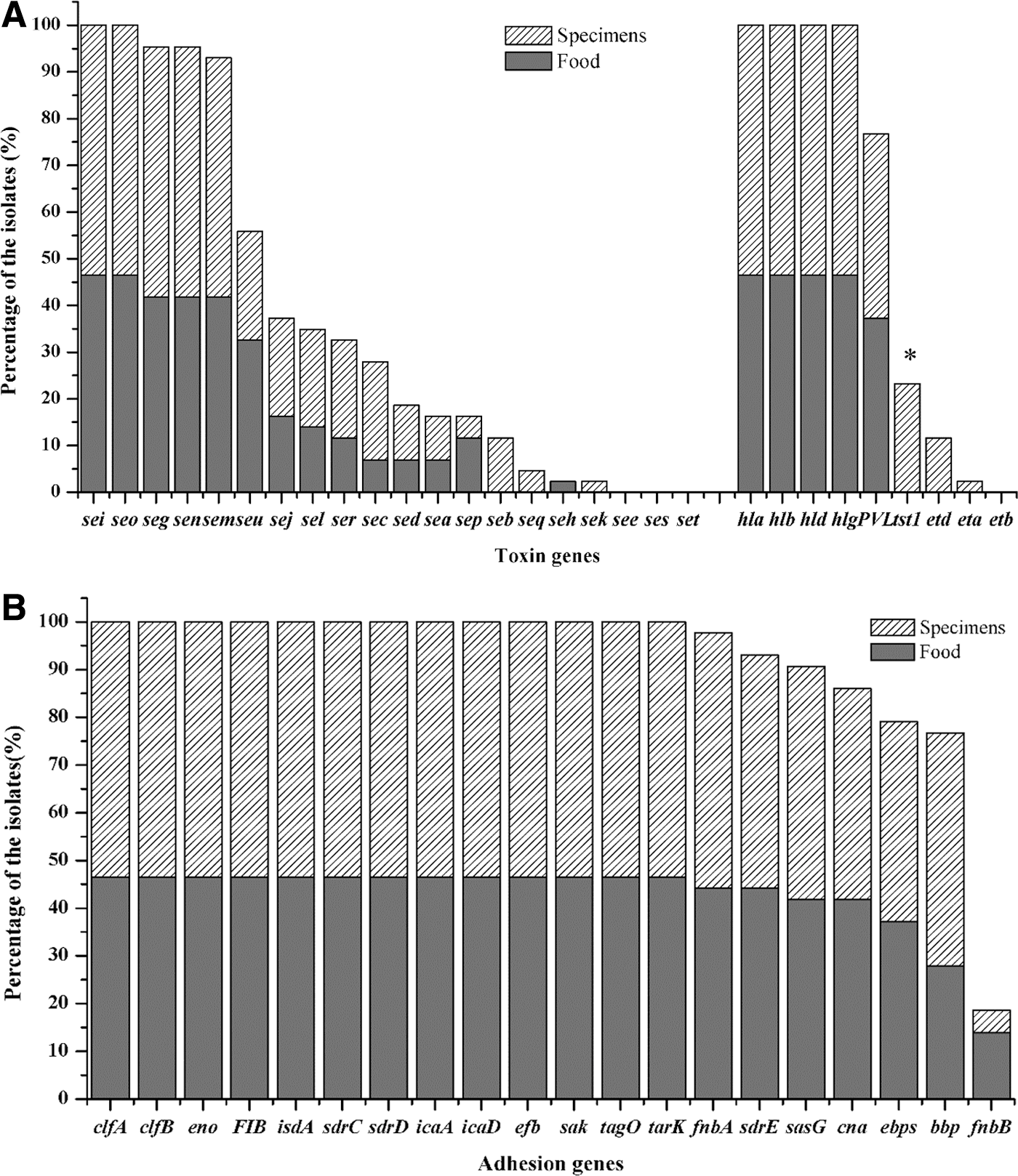
The prevalence of toxin genes
Correlation between virulence gene profiles and the clonality
Toxin gene profiles and its association with MLST and agr types are shown in Figure 4. Forty-three egc-positive isolates displayed 33 different toxin gene profiles, with 8 to 17 toxin genes per isolate. The most prevalent toxin gene profile was egc2-pvl-hla-hlb-hld-hlgv (11.63%), which was found to be associated with CC9-agr II, CC20-agr I, CC72-agr I, and CC121-agr IV. Of the remaining toxin gene profiles, 26 toxin gene profiles were strain specific. Overall, these egc-positive isolates harbored diverse toxin gene profiles and there was no significant association between toxin gene profile and clonality.
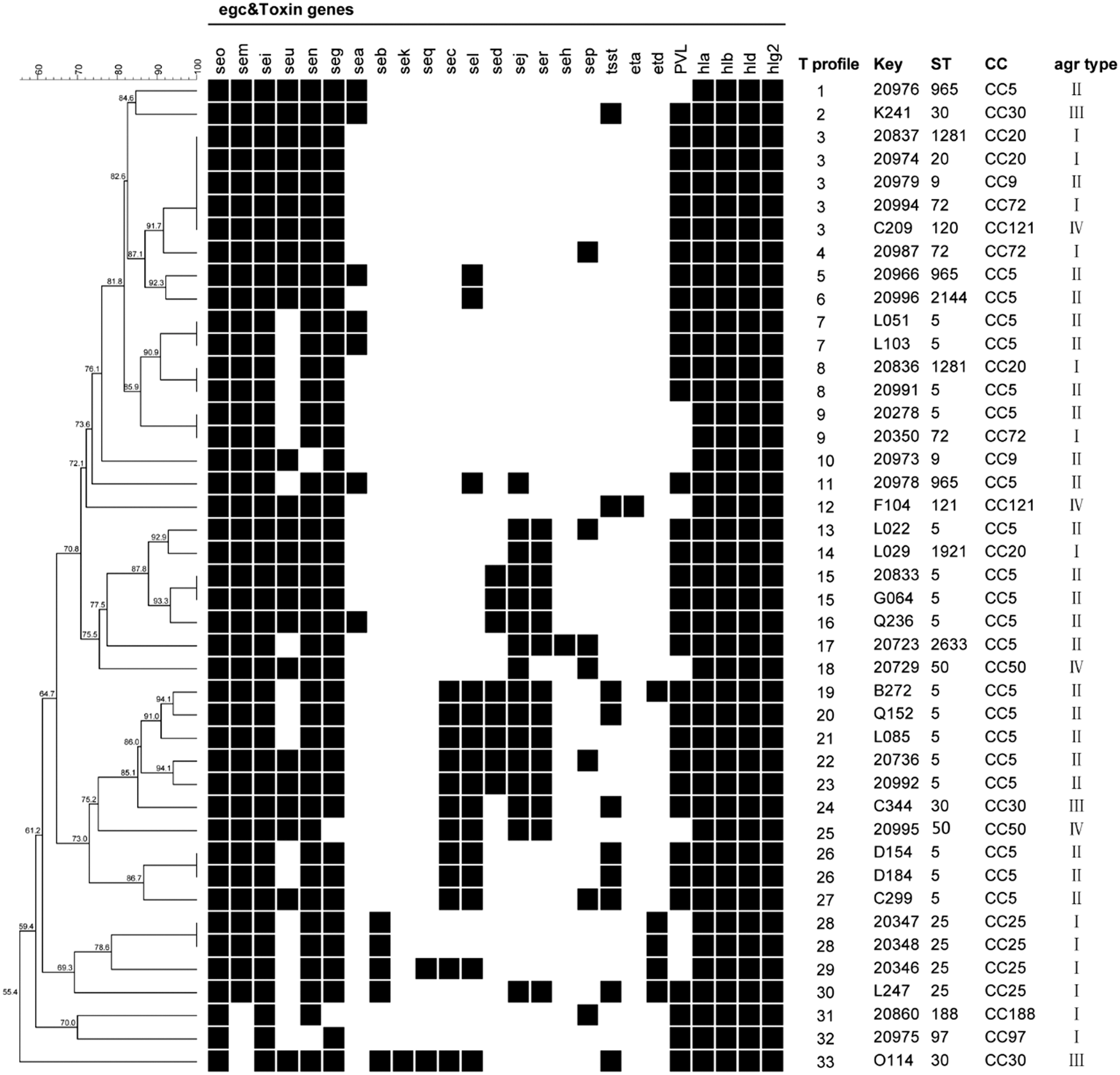
Toxin gene profiles and its association with genotypes of egc-positive isolates. UPGMA dendrogram displaying clustering and genetic similarity (using Jaccard's coefficient) of toxin gene profiles in egc-positive isolates. Toxin genes see, ses, set, and etb were not found in any tested isolates, thus they were not listed in toxin gene profiles. “T profile” means toxin gene profile. Black box, positive; white/blank, negative.
Adhesion gene profiles and its association with clonality were also analyzed (Fig. 5), and it was found that there were 13 adhesion gene profiles in 43 egc-positive isolates. The most prevalent adhesion gene profile was A profile 4 (39.53%), which is associated with CC5-agr II, CC9-agr II, CC30-agr III, and CC97-agr I. Many isolates belonging to the same clonal lineage had a specific adhesion gene profile, such as CC50-agr IV associated with A profile 1, CC121-agr IV associated with A profile 3, CC20-agr I associated with profile 10, and CC72-agr I associated with A profile 11. Moreover, some adhesion genes were also associated with clonal lineages, for example, ebpS absent in CC25-agr I and CC20-agr I, sdrE absent in CC121-agr IV, and fnbB present in CC72-agr I and CC20-agr I. Taken all together, most adhesion genes were conserved and associated with clonal lineage.

Adhesion gene profiles and its association with genotypes of egc-positive isolates. UPGMA dendrogram displaying clustering and genetic similarity (using Jaccard's coefficient) of adhesion gene profiles in egc-positive isolates. “A profile” means an adhesion gene profile. Black box, positive; white/blank, negative.
Discussion
Previous studies have revealed a high frequency of egc genes in the S. aureus population. Mempel et al. (2003) found that 48% of the SE-positive isolates from atopic eczema harbored egc genes, and Zhang et al. (2013) reported that the prevalence of egc genes was 29.5% in isolates from foods in China. Compared with previous results, our data showed a lower frequency of the egc locus in both clinical isolates (18.55%) and isolates from food (14.08%). This difference may be partly related to the geographic origin or isolate source. More importantly, previous studies determined the presence of the egc locus by PCR results for individual SE genes, which may be located outside the egc locus (Vicosa et al., 2013); thus, it may result in a false judgment in the occurrence of the egc locus only based on the detection of individual SE genes. In the present study, egc was identified using a real-time PCR method targeting the highly conserved region of the egc locus (Fusco et al., 2011). Moreover, by combining the PCR detection of egc-borne SE genes, a total of seven different egc profiles were found, which further demonstrated the utility of this real-time PCR assay and its potential for rapid detection of the egc locus.
Our results highlight the diverse genetic background of the egc-positive population by PFGE, MLVA, and MLST. In the present study, PFGE is highly discriminatory, but it may not reflect the actual phylogenetic relationship among egc-positive isolates. For example, two CC9 isolates were separated into two distinct clusters by PFGE. In addition, the MLVA result showed a strong relationship with the MLST result, and MLVA was able to separate egc-positive isolates that were indistinguishable by MLST.
The distribution of the egc locus has been found to be strongly associated with certain clonal lineages (van Belkum et al., 2006). Our results revealed that 10 different clonal lineages and four agr alleles were associated with the egc locus. ST5-agr II, CC25-agr I, and CC30-agr III were mainly identified in clinical isolates in this study. In accordance with our result, Rasmussen et al. (2013) reported that the isolates of CC5, CC25, and CC30 showed an association with invasive disease. In contrast, CC9-agr II and CC97-agr I were found in isolates from food. This is consistent with the observations that CC9 and CC97 are the prevalent livestock clones (Cui et al., 2009; Hata et al., 2010). Furthermore, our study showed that the egc-positive isolates from staphylococcal foodborne illness cases belonged to ST5-agr II, ST25-agr I, and ST72-agr I. Among them, the egc-positive isolates of ST5 and ST72 were negative for any classic enterotoxin (SEA-SEE) that contributes significantly to staphylococcal foodborne illness, further providing evidence that egc-encoding enterotoxins are likely to cause staphylococcal foodborne illness (Johler et al., 2015).
Several virulence factors have been assumed to promote the S. aureus colonization, such as egc locus and sec (Ferry et al., 2005; van Belkum et al., 2006; Nowrouzian et al., 2011). Moreover, beta-hemolysin encoding gene hlb plays an important role in skin colonization (Katayama et al., 2013). In the present study, sec was the most prevalent classic enterotoxin gene and hlb was present in all egc-positive isolates. In addition, several adhesion factors, such as clfB, isdA, WTA, fnbA, and sasG, were also reported to be involved in the S. aureus colonization (Johannessen et al., 2012). Our data showed that these adhesion-associated genes were present in nearly all egc-positive isolates. Overall, these virulence genes associated with colonization showed a high prevalence in the egc-positive population. A combination of these virulence factors may further promote the S. aureus colonization process.
Our results revealed the high prevalence of invasive virulence genes in the egc-positive population. Notably, the genes encoding PVL, associated with severe pneumonia and soft tissue infections, showed a very high prevalence (76.7%) in egc-positive isolates. A previous study has reported that PVL-encoding genes were present with a high prevalence among methicillin-sensitive colonization isolates (Wardyn et al., 2012). Overall, the high prevalence of PVL-encoding genes in colonization isolates further confirmed that S. aureus infections are preceded by commensal colonization (Bogaert et al., 2004). In addition, tsst-1 gene, responsible for toxic shock syndrome, was prevalent in clinical egc-positive isolates, which is consistent with the fact that tsst-1 is typically present in clinical isolates that are associated with important clinical symptoms (Xie et al., 2011). In the present study, four hemolysin genes (hla, hlb, hld, and hlgv ) were present in all egc-positive isolates. Overall, the high prevalence of these virulence factors associated with invasive disease increases the potential risk of egc-positive isolates in human infection.
Our result showed that toxin gene profile was diverse in egc-positive isolates, even in isolates of the same clonal lineage. In accordance with a previous report, there was no significant association between toxin gene profile and clonality of isolates (Machuca et al., 2013). Adhesion-encoding genes are reported to be highly conserved in S. aureus genomes (Rasmussen et al., 2013). This is in accordance with our results that many adhesion genes showed high prevalence in the egc-positive population, except fnbB, which was only found in CC72 and CC20. Peacock et al. (2002) have reported that adhesion genes are ubiquitously carried by different clonal lineages. However, contrary to a previous report, our data showed that the distribution of some adhesion genes was associated with clonal lineages and most isolates of the same clonal lineage had similar adhesion gene profiles. For example, ebpS was absent in CC25-agr I and CC20-agr I, and sdrE was absent in CC121-agr IV. Some adhesion genes have been reported to play a determinant role in bacterial virulence (Barketi-Klai et al., 2011; Rasmussen et al., 2013). Therefore, the absence of some adhesion genes may affect the virulence of some S. aureus clonal lineage.
Conclusions
Our results highlighted the diverse genetic background of the egc-positive population by combining different molecular typing methods. Moreover, MLVA could be used to better understand the molecular epidemiology of the egc-positive population. In light of our results, the high prevalence of invasive virulence genes increases the potential risk of egc-positive isolates in human infection.
Footnotes
Acknowledgments
This study was supported by the National Key Research and Development Program of China (grant number 2016YFD0401102), the Science and Technology Commission of Shanghai Municipality (grant number 14390711900), and the Shanghai Jiao Tong University Agri-X Fund (grant number AgriX2015005).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.