
Abstract
In northwest Poland, 163 blood and 53 fecal samples of wild boars were collected in winter 2012/13 and 2013/14. All blood samples were tested for the presence of hepatitis E virus (HEV) ribonucleic acid (RNA) by two reverse transcription–polymerase chain reaction (RT-PCR) based methods and by anti-HEV IgG enzyme-linked immunosorbent assay (ELISA). About 17.2% of blood samples were seropositive. One-step nested RT-PCR turned out to be too insensitive (11.6% were positive). Therefore a two-step nested RT-PCR was applied where 25.8% of the blood samples were tested positive for HEV RNA. About 50.0% of blood samples positive in ELISA were also positive in two-step nested RT-PCR. The prevalence of HEV RNA in feces was 9.4%. Based on the results of blood (ELISA, PCR) and fecal (PCR) tests, the overall prevalence of HEV in wild boars in northwest Poland was 36.8%. There was no correlation between the ELISA results and the presence of HEV RNA in plasma or in feces. According to the sequencing results of 348 bp PCR products of HEV, there were four different subtypes identified. Reports on the prevalence of HEV in wild boar populations are varying due to different sensitivities of the detection methods. However, this study reveals based on a highly sensitive method that HEV is widely spread in wild boar populations in the northwestern region of Poland and posing a potential risk to the consumer of game meat.
Introduction
H
The main reservoirs of HEV are domestic pigs and wild boars. Additionally, anti-HEV antibodies or HEV RNA were also detected in other animals such as deer, rats, dogs, cats, mongooses, cows, sheep, goats, avian species, rabbits, and horses (Pavio et al., 2010). Over the past few years, investigations were conducted to determine the presence of HEV in wild boar (Adlhoch et al., 2009; Larska et al., 2015), domestic pigs (Huang et al., 2002; Leblance et al., 2007; Wacheck et al., 2012), and products thereof such as raw sausage and liver sausage (Szabo et al., 2015). Pig manure storage facilities were also investigated for the presence of HEV RNA (Kasorndorkbua et al., 2005). In Eastern Europe for example, in Poland, there are only few studies on HEV prevalence and most investigations were based solely on enzyme-linked immunosorbent assay (ELISA) detection methods (Larska et al., 2015). Since sensitivity and specificity of each ELISA kit is variable, additional methods of detection should be applied to validate or to compare the results. Therefore, this study aims to evaluate the sensitivity of methods of detection and the prevalence of HEV (anti-HEV IgG vs. HEV RNA) in blood and in feces of wild boars in the northwestern region of Poland.
The methods of detection were ELISA and two reverse transcription–polymerase chain reaction (RT-PCR) based methods with subsequent nested real-time PCR. RNA is easily degraded by enzyme RNase after sampling. Additionally, depending on stage of infection, RNA may be present only in a low amount in samples such as in blood or in feces. Low levels of RNA may give false negative results when testing with PCR based methods using only a single pair of primers. To minimize this obstacle, nested PCR with two sets of primer pairs is applied, since it increases sensitivity and specificity of quantitative PCR (Haff, 1994). In this study, the external set (3156N-F and 3157N-R) and the internal set (3158N-F and 3159N-R) of primer pairs were modified from Huang et al. (2002). The modification is based on the sequence alignment of the open reading frame (ORF)2 genes of HEV identified in wild boars and in humans in central Europe (mainly in Germany) provided by GenBank NCBI (wild boar: accession no. KF303499.1, KF303501.1, KF303502.2, FJ998008.1, FJ956757.1, FJ998015.1, FJ998019.1; human: accession no. KC618402.1, KC618403.1).
Materials and Methods
Samples
A total of 163 heparin-blood and 53 fecal samples from wild boars were collected in 5 areas in northwestern Poland (Fig. 1). All samples were collected during hunting season (from November to January) in 2012/2013 and 2013/2014. Samples were stored directly after sampling at −20°C until investigation. Directly before testing, heparin blood samples were centrifuged at 3000g for 15 min at 4°C. The supernatants were subjected to ELISA and RNA extraction. Two positive control samples of complementary DNA (cDNA) of HEV were provided from the Friedrich-Loeffler-Institute (FLI), Germany.
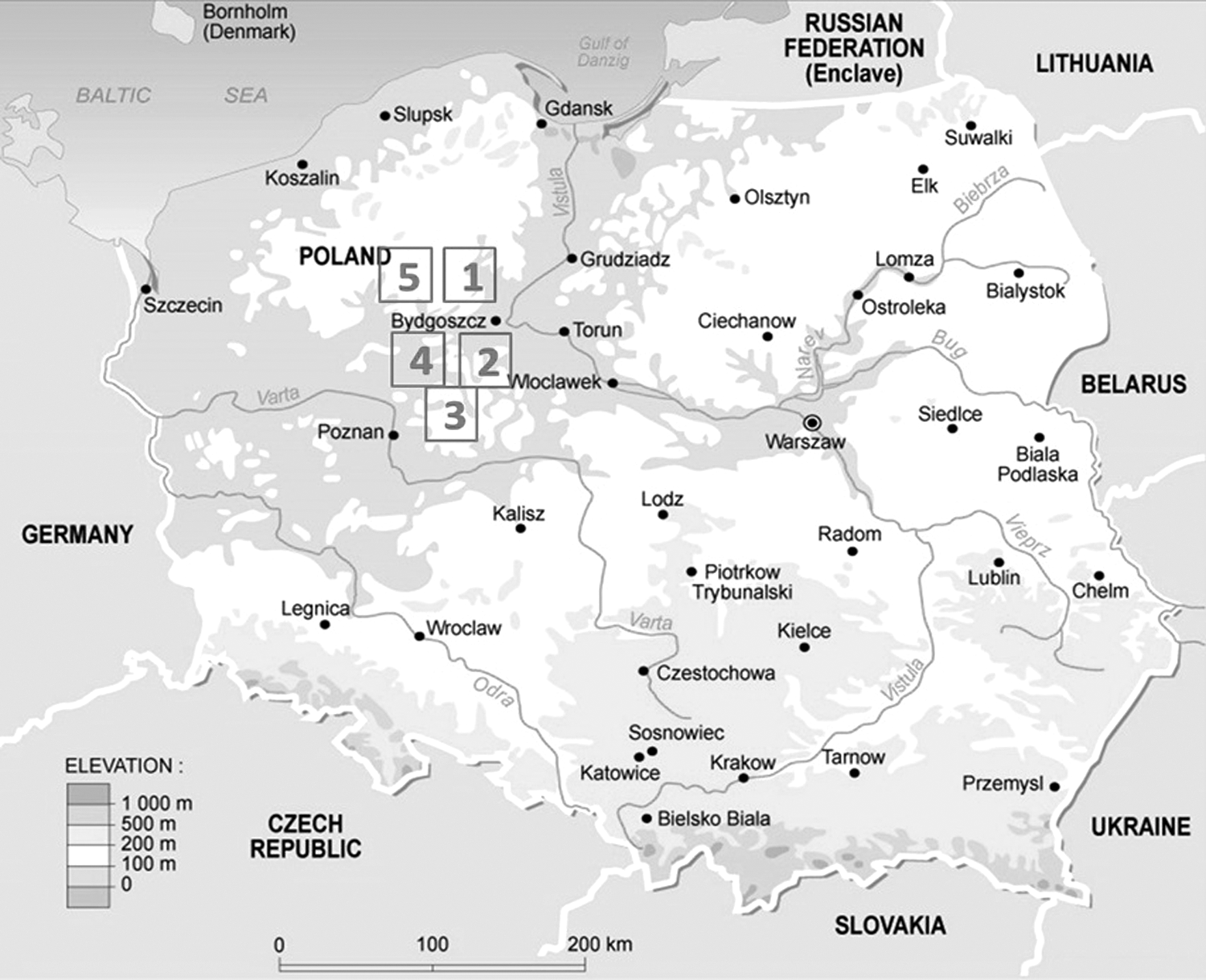
Areas of sampling (1) Sepolno Kraj and its surrounding (Lutowko, Swit, Rozanna, Mrocza, Bory Tucholskie); (2) Miradz; (3) Golabki and Skorzecin; (4) Kcynia and Mierucin; (5) Runowo Kraj.
Enzyme-linked immunosorbent assay
Plasma samples were tested for the presence of anti-HEV IgG of the ORF2 and ORF3 of the genotypes 1 and 3 using the ELISA kit PrioCheck® HEV Ab Porcine kit (Prionics AG, Switzerland). The reaction was read optically in the ELISA reader (Tecan, Switzerland) at a wave length of 450 nm (OD450) with a reference filter at 620 nm. The results were interpreted by following the instructions of the ELISA kit. A cutoff value was calculated as mean OD450 of the cutoff control multiplied with 1.2. Results obtained above or equal to this value were considered positive.
Extraction of RNA
The RNA of HEV from plasma samples was extracted by following the instructions of a commercial kit NucleoSpin® RNA II (Macherey-Nagel, Germany). The RNA extraction kit for fecal samples was RNeasy Mini Kit (Qiagen). Feces of wild boars were first suspended 1:10 in NaCl solution. About 100 μL of plasma and 100 μL fecal suspensions were subjected to RNA extraction. The extracted RNAs were processed directly by one-step and two-step nested RT-PCR described below.
One-step nested RT-PCR
In a single tube, cDNA is synthesized and subsequently amplified in PCR. Single reaction (20 μL) contained 10 μL of 2 × SensiFAST™ SYBR® No-ROX One-Step mix and 0.20 μL reverse transcriptase, 0.4 μL RiboSafe RNase Inhibitor (Bioline, Germany), 3.6 μL H2O, 5.0 μL extracted RNA template, and 0.4 μL of each 20 μM primer (external primer sets: 3156N-F: AAT TAY GCC CAG TAT CGG GTY G and 3157N-R: CCT TRT CYT GCT GHG CAT TCT C). The one-step RT-PCR was performed by Biorad CFX96 Touch™ (Biorad) with following steps: 15 min at 45°C for reverse transcription, followed by initial denaturation 5 min at 95°C, and 40 cycles of 5 s at 95°C, annealing 10 s at 60°C, and elongation 20 s at 72°C. Melting curve analysis was performed at the end of the real-time PCR program. The PCR product was diluted 1:3 in RNAse-free water. The diluted PCR product served as template for the second real-time PCR run using the internal primer set (3158N-F: GTW ATG CTT TGY ATY CAT GGC T and 3159N-R: AGC CGA CGA AAT YAA TTC TGT C). In 20 μL reaction mixture contained 10 μL of 2 × SensiFAST SYBR No-ROX mix (Bioline, Germany), 0.4 μL of each 20 μM primer (final concentration 400 nM), 4.2 μL H2O, and 5.0 μL DNA template. The real-time PCR program was as following: initial denaturation 5 min at 95°C, followed by 40 cycles of 5 s at 95°C, annealing 10 s at 56°C, and elongation 15 s at 72°C. Additionally, the product of the second real-time PCR run was loaded in 1% agarose gel.
Two-step nested RT-PCR
For two-step nested RT-PCR, the conversion of RNA into cDNA was performed according to the instruction of QuantiTect® Reverse Transcription Kit (Qiagen). To eliminate genomic DNA, 12 μL of eluted RNA was mixed with 2 μL gDNA Wipeout Buffer (7 × ) and incubated at 42°C for 2 min. The entire volume from the first step (14 μL) was mixed with 1 μL reverse transcriptase, 4 μL RT-buffer, and 1 μL RT-random primer. The entire mixture (20 μL) was incubated at 42°C for 45 min. After that, the mixture was incubated at 95°C for 3 min to inactivate the enzyme Quantiscript Reverse Transcriptase. The cDNA was stored at −20°C until testing by nested PCR.
The first real-time PCR run was done by using the external primer set 3156N/3157N. Single reaction (20 μL) contained 10 μL of 2 × SensiFAST SYBR No-ROX mix (Bioline, Germany), 0.4 μL of each 20 μM primer, 4.2 μL H2O, and 5.0 μL cDNA template. The real-time PCR step was as following: initial denaturation 5 min at 95°C, and 40 cycles of 5 s at 95°C, annealing 10 s at 60°C, and elongation 20 s at 72°C. The dilution of PCR products, reaction mix and PCR conditions of the second real-time PCR run, and proof of the fragment in 1% agarose gel were as described in section one-step nested RT-PCR. PCR products with the expected fragment of ca. about 348 bp were purified using the MiniElute Kit (Qiagen) and submitted to Eurofins, Germany (
Sensitivity of one-step and two-step nested RT-PCR
To demonstrate the sensitivity of one-step and two-step nested RT-PCR, 10-fold dilutions of four positive samples (three blood and one fecal samples) from 10−1 to 10−4 was performed. Samples were diluted in RNA extract from HEV-negative blood and fecal sample, since the matrix type may influence PCR amplification. All dilutions were tested with one-step and with two-step nested RT-PCR.
Statistical analysis
The correlation between ELISA and RT-PCR results was analyzed using correlation coefficient statistical method (Excel software).
Results and Discussion
One-step and two-step nested RT-PCR
One-step RT-PCR requires specific primer pairs and generally less handling steps. Additionally, one-step RT-PCR is also reported to achieve higher (or at least the same) sensitivity level, compared to two-step RT-PCR (Wacker and Godard, 2005; Picard-Meyer et al., 2015). In this study, by using the one-step nested RT-PCR method, HEV RNA was detected in 11.6% of blood samples while tested with two-step nested RT-PCR, 25.8% of samples were positive. A similar result was reported by Al-Shanti et al. (2009). Sellner and Turbett (1998) also found that the two-step (uncoupled) RT-PCR based method has a higher sensitivity than one-step (coupled) RT-PCR, particularly if the concentration of target RNA in the sample is very low. A low amount of RNA in samples may be according to the stage of HEV infection. Additionally, RNA is easily degraded by enzyme RNase, if samples are not properly handled after sampling (Cattoli and Monne, 2009).
To support this result, a tenfold dilution series (up to 10−4) of RNA extracts from four positive samples was tested with both one-step and two-step RT-PCR methods. Figure 2 shows amplified fragments of the second PCR run with primer pair 3158N-F and 3159N-R. The dilution 10−3 of blood sample 1 and 2 were positive with two-step nested RT-PCR, while the one-step nested RT-PCR was negative. In this test, the sensitivity of two-step RT-PCR was slightly higher than the sensitivity of one-step RT-PCR, namely up to 1 log10 level of the RNA extract dilutions. However, the sensitivity of one-step RT-PCR can be optimized with proper PCR conditions. Additionally, each PCR kit and each primer pair should be optimized and validated before applying because PCR inhibitors may be present in different types of sample matrices, or even different types of PCR instruments may bias the results (Roux, 2009; Buckwalter et al., 2014; Picard-Meyer et al., 2015).

Sensitivity of one-step versus two-step RT-PCR. (−): negative control, (+): positive control: cDNA from Friedrich-Loeffler-Institute; B1, B2, B3, and F are pure RNA extracts from three blood and one fecal samples; −1, −2, −3, and −4 are 10-fold serial dilution (10−1, 10−2, 10−3, and 10−4) of RNA extracts. RT-PCR, reverse transcription–polymerase chain reaction; cDNA, complementary DNA.
The specificity of both RT-PCR methods depends on the primers used. Additionally, nested PCR with two sets of primer pairs increases both sensitivity and specificity of the test. In this study, the specificity of applied primer pairs was 100%, since all positive samples show only similarity to sequences of HEV provided in GenBank (see Section Phylogeny of sequenced HEV RNA).
Enzyme-linked immunosorbent assay
Figure 3 shows the prevalence of HEV (ELISA vs. two-step nested RT-PCR) in blood samples of wild boars considering the year and the sampling area. In total 163 blood samples of wild boars were collected in five areas in northwestern Poland in winter 2012/13 and 2013/14. The seroprevalence of anti-HEV IgG of wild boars sampled in winter 2012/13 was 19.1% (21/110) and 13.2% (7/53) in 2013/14, with an overall prevalence of 17.2% (28/163).

% HEV positive (ELISA vs. two-step nested RT-PCR) blood and fecal samples of wild boars considering the year and sampling area in northwestern Poland (Fig. 1). HEV, hepatitis E virus; RT-PCR, reverse transcription–polymerase chain reaction; ELISA, enzyme-linked immunosorbent assay.
Correlation of anti-HEV IgG and HEV RNA in blood and in feces
The prevalence of HEV based on the detection of HEV RNA in blood using two-step nested RT-PCR was also higher than the prevalence of anti-HEV IgG (Fig. 3), namely 30.0% (33/110) in winter 2012/2013 and 17.0% (9/53) in winter 2013/2014, with an overall prevalence of 25.8% (42/163). In two sampling areas (area 1 and 4), the prevalence of HEV was increasing in 2013/2014 according to the result either of PCR or of ELISA. For fecal samples, 9.4% (5/53) were positive by PCR. One positive fecal sample was from area 2 (Miradz) and four positive fecal samples were from area 3 (Golabki and Skorzecin). Blood samples of wild boars with positive fecal samples were negative for anti-HEV IgG and HEV RNA. Based on the results of blood (ELISA, PCR) and fecal (PCR) test, the overall prevalence of HEV in wild boars in northwestern Poland was 36.8% (60/163).
The sensitivity and specificity of ELISA kits is variable and may alter the results of the test (Adlhoch et al., 2009; Wedemeyer et al., 2012; Dremsek et al., 2013). In this study, the seroprevalence of HEV tested with the ELISA kit PrioCheck HEV Ab Porcine is lower than in a previous study by Larska et al. (2015), in which the seroprevalence of HEV of wild boars of the same region in Poland during 2012–2013 was 68% −88%, using the ELISA kit IDVet Diagnostics (France). In the federal state of Brandenburg of Germany, which borders to northwestern Poland, the seroprevalence of anti-HEV IgG in wild boars was in average 17.2%, tested with ELISA kits from Genelabs Diagnostics (France) and recomWell HEV, Mikrogen (Germany). From the same wild boars, 6% of blood and 100% of the liver samples were positive for HEV RNA (Adlhoch et al., 2009).
HEV RNA circulation in wild boars in northwestern Poland was found in four sampling areas (areas, 1, 2, 3, and 5). In area 4 (Kcynia and Mierucin), no sample was positive for HEV RNA (Fig. 3). In total, 28.8% (47/163) of wild boars have viral RNA in blood or in feces. About 20.2% (33/163) were probably new infections since the seroconversion of anti-HEV IgG was not detected by ELISA (Wedemeyer et al., 2012). Five wild boars with presence of HEV RNA in feces are potentially shedding HEV into the environment. One week after infection, pigs can shed virus in feces in large amounts for 3–4 weeks, while viremia may be transient or absent (Kasorndorkbua et al., 2004). The other wild boars can accidentally be infected by consumption of HEV contaminated feed and water.
Table 1 shows the correlation between blood samples positive for anti-HEV IgG and blood samples positive for HEV RNA. About 50.0% (14/28) of the ELISA-positive samples were also positive in PCR. About 66.7% (28/43) samples positive in PCR were negative in ELISA. There was no correlation between the ELISA result and the presence of HEV RNA in plasma or in feces in this study. Similar observation was reported by Leblance et al. (2007) and Adlhoch et al. (2009). According to Ahmed et al. (2015), the correlation between anti-HEV antibodies and the presence of HEV RNA is relatively low. This depends on the stage of HEV infection, in which either antibodies or RNA is present. Additionally, the sensitivity and specificity of ELISA kits, PCR based methods, and primers used may lead to varying results.
HEV, hepatitis E virus; RT-PCR, reverse transcription–polymerase chain reaction; ELISA, enzyme-linked immunosorbent assay; RNA, ribonucleic acid.
Phylogeny of sequenced HEV RNA
PCR products of two-step nested RT-PCR from 42 blood and 5 fecal samples with an expected fragment of ca 348 bp were sequenced. All sequences were part of ORF2 genes of HEV genotype 3. There were four different subtypes identified. The majority of sequences is grouped in subtype B (n = 21) and subtype A (n = 15), followed by subtype D (n = 7) and subtype C (n = 4). All subtypes show 91–94% identity to the sequence provided in GenBank (accession no. KR027387.1: human, France; JQ424440.1: pigs, Bolivia; AB986280.1: human, Japan/KF719309.1: pigs, USA and KR027293.1: human, France, respectively). Figure 4 shows phylogeny of 47 sequences (four subtypes) of HEV RNA from wild boars in northwestern Poland compared with sequences of HEV from GenBank (NCBI), which were found in wild boars, domestic pigs, and humans in Europe, USA, Japan, and Bolivia.

Phylogeny of 47 sequences of HEV RNA from wild boars in northwestern Poland and similar sequences of HEV found in Europe, USA, Japan, and Bolivia. HEV, hepatitis E virus.
Conclusions
The prevalence of HEV in blood samples of wild boars in northwest Poland was 25.8% tested with two-step nested RT-PCR, 11.6% with one-step nested RT-PCR and 17.2% with ELISA. As a result, methods of detection of HEV RNA in biological products should be considered. A low amount of RNA may lead to false negative results when using a method of detection with low sensitivity. By using two-step RT-PCR based methods and subsequent nested real-time PCR in this study, a high number of wild boars was found positive for HEV RNA. This might pose a risk to exposed person like hunters, meat processing staff, or consumer of wild game.
Footnotes
Acknowledgments
We would like to thank the technical assistants from the Chair of Food Safety, Faculty of Veterinary Medicine, LMU Munich, Germany for their help.
Disclosure Statement
No competing financial interests exist.