
Abstract
Introduction:
Pathogenic Escherichia coli has been listed among the most important bacteria associated with foodborne illnesses around the world. We investigated the genetic relatedness among Shiga toxin–producing E. coli (STEC) isolated along the animal food supply chain and from humans diagnosed with gastroenteritis in Qatar.
Methods:
Samples were collected from different sources along the food supply chain and from patients admitted to the hospital with complaints of gastroenteritis. All samples were screened for the presence of E. coli O157:H7 and non-O157 STEC using a combination of bacterial enrichment and molecular detection techniques. A proportional sampling approach was used to select positive samples from each source for further multilocus sequence typing (MLST) analysis. Seven housekeeping genes described for STEC were amplified by polymerase chain reaction, sequenced, and analyzed by MLST. Isolates were characterized by allele composition, sequence type (ST) and assessed for epidemiologic relationship within and among different sources. Nei's genetic distance was calculated at the allele level between sample pools in each site downstream.
Results:
E. coli O157:H7 occurred at a higher rate in slaughterhouse and retail samples than at the farm or in humans in our sampling. The ST171, an ST common to enterotoxigenic E. coli and atypical enteropathogenic E. coli, was the most common ST (15%) in the food supply chain. None of the genetic distances among the different sources was statistically significant.
Conclusion:
Enterohemorrhagic E. coli pathogenic strains are present along the supply chain at different levels and with varying relatedness. Clinical isolates were the most diverse, as expected, considering the polyclonal diversity in the human microbiota. The high occurrence of these food adulterants among the farm products suggests that implementation of sanitary measures at that level might reduce the risk of human exposure.
Introduction
T
When parsing out the causative factors of the global burden of foodborne illness, strains of enterohemorrhagic Escherichia coli (EHEC) are notable for causing particularly serious bouts of foodborne illness (Hoffmann et al., 2012). These pathogenic infections are also increasing, with a 15% rise in EHEC cases among European countries between 2000 and 2004 (Gil et al., 2004). The EHEC pathotype can be defined as the following: possessing the eae gene with stx1 gene, stx2 gene, or both (Bletz et al., 2013; Kotloff et al., 2013). The eae gene codes for intimin adherence protein and is required for intimate attachment to epithelial cells (Jerse et al., 1990). The stx1 and stx2 genes both code for Shiga-like toxin, an enterotoxin that inhibits the eukaryotic 60S ribosomal subunit and prevents protein translation (O'Loughlin and Robins-Browne, 2001). The most commonly studied serotype of EHEC is O157:H7, responsible for several foodborne illness outbreaks, including a high-profile 1993 U.S. outbreak (Kotloff et al., 2013). However, non-O157:H7 EHEC serogroups have been reported and these bacteria are capable of causing severe diarrheal disease (Shen et al., 2015). Furthermore, their prevalence has sharply risen by 51.7% in European countries between 2001 and 2004 (Hoffmann et al., 2012). Other studies have shown that the prevalence of non-O157 EHEC isolates may be similar or even superior to O157:H7 prevalence in clinical cases (Feng et al., 2010). Despite the increase, the impact of non-O157:H7 EHEC on the risk of gastroenteritis is still poorly understood.
EHEC-caused foodborne illness begins with severe diarrhea and in many cases progresses to bloody diarrhea (Bryan et al., 2015). In addition to severe diarrhea, other symptoms are typical of foodborne illness: fever, nausea, and abdominal cramps (Bryan et al., 2015). Approximately 10% of those affected will develop hemolytic uremic syndrome (HUS), characterized by renal failure, hemolytic anemia, and thrombocytopenia (Bryan et al., 2015). HUS is the leading cause of acute renal failure in children (Bryan et al., 2015). Currently, treatment for HUS is only supportive in nature (Bryan et al., 2015). Several routes have been incriminated in the likelihood of transmission of these pathogens (Kintz et al., 2017). Therefore, a reasonable approach to mitigate the associated risk of infection with these pathogens is to minimize contamination of the food supply chain and prevent exposure of food-producing animals and humans.
Therefore, investigating the threats of these pathogens to the food supply system would provide useful information that could be used to mitigate their potential risk (Duffy et al., 2008; Alexander et al., 2010; Monaghan et al., 2012). Multilocus sequence typing (MLST) is a powerful molecular technique in investigating the genetic relatedness among bacterial isolates (Maiden et al., 1998). It involves polymerase chain reaction (PCR) amplification of seven highly conserved housekeeping genes, and allelic variants in these conserved genes can be uncovered through the sequencing of the PCR products, providing an unambiguous data source to classify and characterize the bacteria (Urwin et al., 2003). The unique seven allele combination then comprises a unique sequence type (ST), which provides the means to investigate the relatedness among isolates recovered from different sources and identifies possible virulence profiles. The combination of epidemiologic and molecular data allows relationships within the supply chain to be both quantified and assessed for impact on human health (Urwin et al., 2003; Jaros et al., 2016).
This study was planned to explore the genetic relatedness among EHEC isolates from the food supply chain of animal origin and in patients admitted with gastroenteritis in a highly dynamic population, Qatar. The long-term hope is to identify critical control points for intervention along the food supply chain.
Materials and Methods
Source of samples
Samples were collected using a random sampling scheme by generating a list of random numbers using Excel, at three different production levels and from humans admitted to the hospital with the complaint of gastroenteritis. Detailed descriptions of the samples and the sampling are provided elsewhere (Mohammed et al., 2015). The number and source of samples that were collected and tested are shown in Table 1. In brief, the food supply chain sources included animals on farms (cows, camel, and sheep), abattoir samples included carcass swabs after processing, and retail samples included processed raw animal products at retail. All livestock samples were collected from apparently healthy animals. Fecal samples and udder swabs were collected from farms. Carcass swabs were collected from dressed animals at the abattoir. A letter of solicitation was sent to all owners/farm mangers explaining the objectives of the study and samples were obtained from consenting owners. The samples were screened for the presence of E. coli serogroups (Shiga-producing O157:H7 and non-O157 EHEC) using a combination of bacterial enrichment and molecular detection (Mohammed et al., 2015).
Number tested positive out of all the samples collected.
Human samples were obtained from individuals who were admitted with complaints of gastroenteritis to any of the Hamad Medical Corporation (HMC) hospitals in Qatar. Fecal samples were collected and tested bacteriologically for E. coli. Gastroenteritis cases consisted of individuals of varying ethnicity, nationality, age, gender, and received clinical diagnosis. The samples were collected as routine patient care with ethical approval from the institutional review board to use the samples and data. Although the initial isolation was performed at the HMC bacteriological laboratory, the results were confirmed using the molecular technique applied to animal and retail samples (Mohammed et al., 2015).
Culturing and genotyping
A proportional sampling technique based on the source of the sample was used to select EHEC-positive samples (serogroup/stx/eae) for further genetic analyses to address the objectives of this study.
Selected samples were cultured into the brain–heart infusion broth overnight in a 37°C incubator. The cultured samples were then streaked on MacConkey agar; a single colony matching the E. coli morphology was selected for genomic DNA extraction. The single colony was streaked again on MacConkey agar repeatedly, four times, in a dilution pattern onto the agar. A single colony was picked from the final dilution of the streaking, and conformed to its original type, using the Bax PCR before extraction of the DNA. iGenomic DNA was isolated from this single colony of E. coli using the MasterPure DNA Purification Kit (Epicentre Biotechnologies, Madison, WI). A total of seven housekeeping genes (aspC, clpX, fadD, icdA, lysP, mdh, and uidA) were selected and used for PCR amplification in the MLST scheme. The primer sequences and PCR conditions used were obtained from the MLST of pathogenic E. coli database available at (
PCRs were set up with 1 μL of 50–100 ng/μL DNA template, 2.5 μL of 2 mM deoxynucleoside triphosphates (dNTP mix), 2.5 μL of 10 × DreamTaq Buffer (includes 20 mM MgCl2) (Thermo Scientific, Fermentas), 1 μL of 10 μM primer pairs, and 0.3 μL High Fidelity Taq DNA Polymerase 5 U/μL (DreamTaq; Thermo Scientific, Fermentas). PCR products were then purified using the ExoSAP-IT purification kit (Affymetrix, Inc., Santa Clara, CA). DNA sequencing was performed at the Cornell University Biotechnology Resource Center using the Applied Biosystems Automated 3730 DNA Analyzer (Thermo Fisher Scientific).
Genetic and statistical analyses
The sequence for each gene was assembled and proofread using CodonCode Aligner (CodonCode Corporation, Centerville, MA). All sequences were manually examined to confirm the sequence and correct for chromatogram overlap. The aligned sequence contig of each individual gene was submitted to the MLST database website (
After generating the allele and ST designations, BioNumerics v.7 was used to generate a minimum spanning tree for categorical data. Nei's genetic distance was calculated at the allele level, yielding a value ranging from 0 (when the two groups have no genotypes in common) to 1 (when the two groups have the same genotype distribution) (Rotariu et al., 2009). The statistical significance of the genetic distance was determined through an Excel Visual Basic Application (VBAE) by comparing the genetic distance with a distribution of 10,000 genetic distances generated by randomizing the data without replacement (Manly, 1985, 2007).
Descriptive statistics and assessment of the significance of associations between the sources of the sample were performed using SPSS v23 (IBM-Statistical Software, White Plain, NY). The likelihood of isolating a particular pathotype from a specific source was quantified using an odds ratio (OR). Significance of association between the presence of the pathogens and the source of sample was evaluated using the chi-square and Fisher's exact tests. Correlation among sources of the samples was computed using the Spearman test. Significance was considered at p-values <0.05.
Results
Distribution of samples by source
A total of 100 samples were selected from all the positive samples identified in the study. Table 1 shows the distribution of the selected samples by source, including the total collected and total positive. Table 1 shows the number of samples selected by source, proportionally to the total positive. Fifty percent of the selected samples were from animals at the farms, whereas 30% of the samples were from human cases with gastroenteritis. It was three times more likely to isolate E. coli O157:H7 from animals at the production site (farm) than from cases with gastroenteritis (OR = 3.4, 95% confidence interval [0.9–13.4], p = 0.05). It was also more likely to isolate E. coli O157:H7 from slaughterhouse or retail, in comparison to humans with gastroenteritis. However, the association was not significant due to the large variability among the sources.
The results of the samples that were selected for sequencing are shown in Table 1. The stx1, stx2, and eae genes were detected in all the samples from farms, slaughterhouses, and retail (Table 1). The stx and eae genes were detected in samples collected from gastroenteritis cases at the rates of 7% and 37%, respectively. All the selected samples for sequencing were screened for the six food adulterants of non-O157 E. coli (Table 2). While all the farm samples belonged to one of the six serogroups, only 7%, 6%, and 13% isolates from other sources (slaughterhouse, retail, and humans, respectively) were identified to belong to one of these non-O157 food adulterant serogroups. The majority of the farm samples belonged to serogroup O103 and O26 (28% and 24%, respectively). Serogroups O26 and O45 were detected in all of four sample sources, but farm samples had the highest prevalence, 75% and 61%, respectively. Serogroups O121, O103, and O145 were not detected in samples collected from human cases of gastroenteritis. Since the majority of the samples were from farm animals (50 samples) and from cases of gastroenteritis (30 samples), we compared the proportions of these serogroups between those two sources (Table 3). Serogroups O26, O121, and O103 were detected at a significantly higher proportion among farm samples in comparison to human samples (0.24, 95% CI 0.13–0.35 vs. 0.05, 95% CI 0.0–0.15 and 0.16, 95% CI 0.19–0.44 vs. 0, 95% CI 0–0.15) (Table 3). Serogroup O111 was detected at a significantly higher proportion among the human samples in comparison to the farm samples (Table 3). The correlation of the presence among these serogroups from farm and from humans was assessed using Spearman ranked correlation, and the only relatively common serogroup between farm sources and humans was O45 (correlation coefficient was 0.43; data are not shown).
MLST analysis of EHEC isolates
Among the 61 isolates identified as EHEC with complete sequencing, 38 different STs were found by MLST analysis. The most common ST was 171 (15%), followed by 303 (7%), 27 (7%), and 243 (7%) (Table 4). These STs were all found in human samples (Table 4). Among these STs, 20 (51%) had never been previously reported. Of these 20 new STs, 19 were singletons in the data set (Fig. 1). The study identified one novel aspC allele, two new fadD alleles, two new icdA alleles, and one new uidA allele (Table 4).
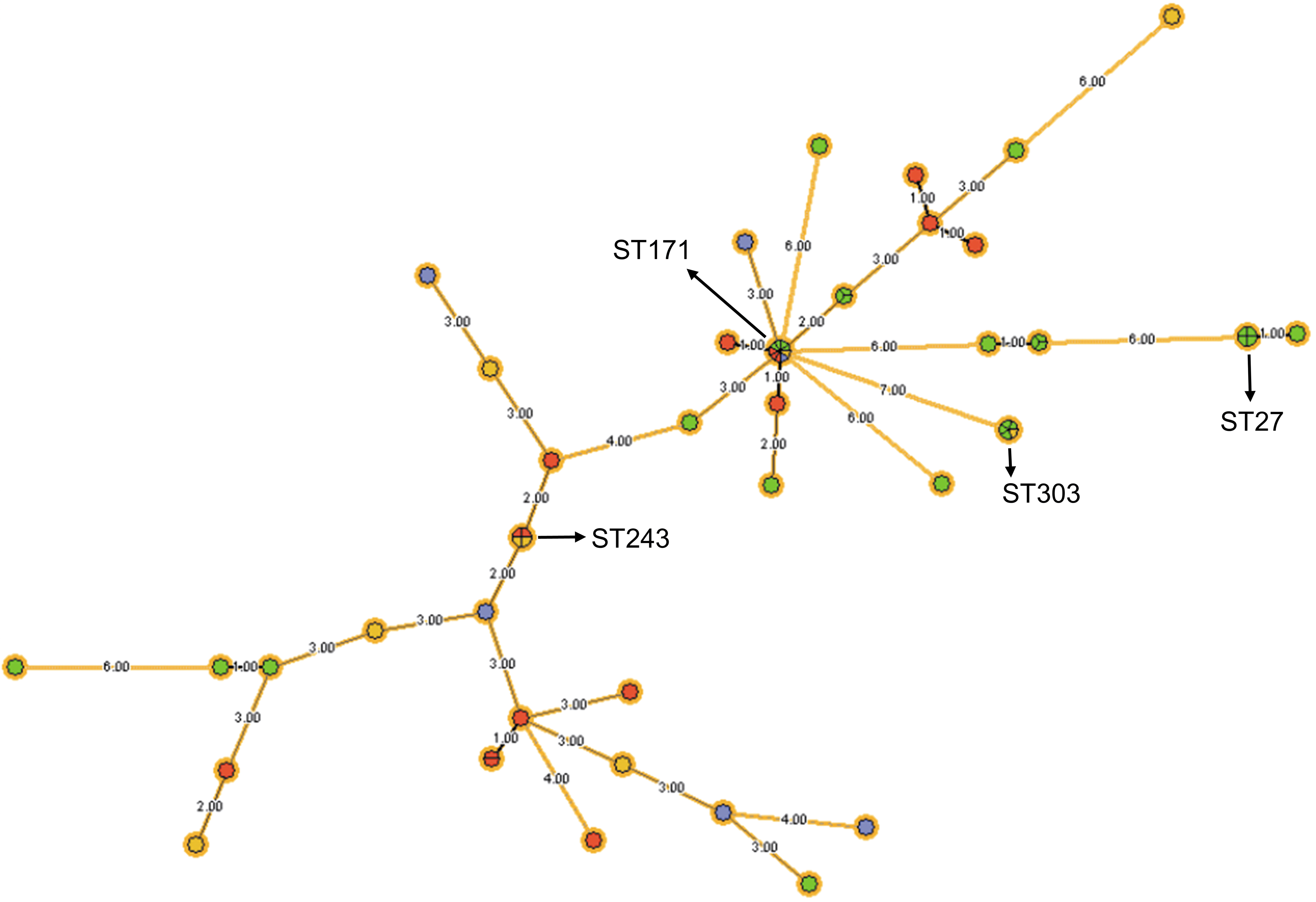
Minimum spanning tree demonstrating the multilocus sequence typing patterns of 61 confirmed EHEC isolates from four different sources, to evaluate genetic relatedness of the isolates using the “farm-to-fork” concept. Each node represents one ST. Farm isolates in yellow, slaughterhouse isolates in red, retail isolates in blue, and human isolates in green. Most prevalent STs are indicated as numbers. The distances (in terms of number of locus variants) are indicated in the branches connecting the ST nodes. EHEC, enterohemorrhagic Escherichia coli; ST, sequence type.
Genetic diversity among sample pools
Genetic relationship was assessed through a minimum spanning tree for categorical data (Fig. 1). The maximum distance between two points within a sample pool was summed to quantify the genetic diversity within a pool. Retail samples showed the least diversity with a maximum distance of 23, followed by slaughterhouse samples with 25, farm samples with 36, and human samples with 38. To compare genetic overlap among sample pools, Nei's genetic distance was calculated at the allele level between sample pools one site downstream in the supply chain. None of the genetic distances was found to be statistically significant (Table 5). Therefore, EHEC isolates collected from a source along the pathway are genetically homogenous in comparison to isolates collected one site downstream.
ns, statistical difference was not significant (p > 0.05).
Discussion
MLST was chosen to investigate the genetic relatedness among the isolates because its targeted and sensitive approach combines advances in high-throughput sequencing and bioinformatics with established population genetics techniques (Maiden et al., 1998). MLST has utility in epidemiologic investigations—the drawing of comparative conclusions from global data that often comes from wildly different sources (Maiden et al., 1998; Urwin et al., 2003). Furthermore, MLST schemes provide a common, easily reproducible scheme to characterize bacteria from a myriad of sources (Urwin et al., 2003). Therefore, the technique provides information that could be used to mitigate the associated risk with these pathogens.
The study revealed a diverse range of STs with 61 E. coli isolates spread broadly into 38 STs, many of which were singletons. This diversity is possibly facilitated by heavy immigration to Qatar. Qatar is home to a large expatriate population, where expatriates outnumber Qataris almost seven to one (Berrebi et al., 2009). Qatar also imports almost 90% of its food and food-producing animals from different countries (Moghazy et al., 2013). Therefore, beyond the polyclonal nature of the intestinal microbiota, the international food trade and a large global population could also contribute for the observed genetic diversity.
To the best of our knowledge, this study is the first to report the higher prevalence of ST171 among EHEC isolates. However, this ST has been found widely across other E. coli pathotypes and was the most prevalent ST among enterotoxigenic E. coli (ETEC) clinical isolates from Korea (Oh et al., 2014). This ST was also the most prevalent among porcine ETEC isolates from Michigan and a pool of ETEC human isolates obtained from Guinea-Bissau, Saudi Arabia/Egypt, India, Bangladesh, Vietnam, Brazil, United States, Argentina, Senegal, Thailand, Turkey, and Peru (Steinsland et al., 2010).
ST171 was also found in atypical enteropathogenic E. coli (EPEC) O157: non-H7 isolates from water, meat, and clinical samples in the United States, as well as meat samples from France (Feng et al., 2010). Furthermore, ST171 was also found in atypical EPEC O157: H7 strains isolated from dogs in Argentina, clinical samples in Germany, environmental samples in the Netherlands, and clinical samples in Norway (Feng et al., 2012). A Japanese clinical isolate of unreported pathotype and previously undocumented fluoroquinolone resistance was also determined to be ST171 (Sato et al., 2011). Our study adds ST171 EHEC isolates from Qatari farms, slaughterhouses, and retail establishments. The majority of the literature on ST171 shows that this particular ST is among many different pathotypes of infective E. coli globally and emphasizes its zoonotic potential. One of the limitations of this study is that MacConkey is not a selective media for Shiga toxin–producing E. coli.
When analyzing the amount of genetic diversity within each sample pool, the genetic diversity was expected to increase at each point in the supply chain, as EHEC species survive easily along the supply chain. EHEC species can cycle through the low pH of farm ruminants' stomachs and can survive for months in feces or soil (Bolton et al., 1999). Animal water supplies may also be contaminated by biofilm build-up on drinking troughs (Lejeune et al., 2001). EHEC samples also easily survive the stress of food processing conditions (Palumbo et al., 1995). Therefore, there would be a new potential source of pathogens at each new site, as well as the pathogens “carrying-over” from the previous sites in the supply chain. A speculative explanation of low diversity among retail samples could be due to either the low number of isolates or minimal food processing at the retail establishments. Slaughterhouse and farm samples also did not conform to the expectations, as farm samples were shown to be more diverse than slaughterhouse samples. Sanitary measures implemented at the slaughterhouse might decrease bacterial load and hence the genetic diversity of pathogens (Anonymous, 1967). A similar phenomenon may be present in Qatar. Conversely, clinical isolates were the most diverse, which could be attributed to the diversity in the population. Another MLST study also found clinical isolates of Campylobacter to be more diverse than animal samples, most likely due to the variety of pathogenic sources that can infect humans (Gormley et al., 2008). We wanted to forewarn the reader that we have a limited or relatively smaller number of samples from these sources and this might bias the findings in our study.
A hierarchical pathway was assumed by which this pathogen enters the food supply chain—farm→slaughterhouse→retail→humans. The Nei's genetic distances, calculated from EHEC isolates collected at these nodes, were not significantly different. This result fails to preclude the possibility of EHEC pathogenic infiltration and carry-over between farms and slaughterhouses, slaughterhouses and retail establishments, and retail establishments to patients. However, the specific point of any possible infiltration and directionality cannot be determined from this study because the index value was based on the seven housekeeping genes targeted in this study. Further investigation is necessary.
Many studies use MLST to characterize E. coli samples phylogenetically. In addition, these studies also focus on the identification of the infection reservoir, rather than the route of human infection. Extensive MLST studies have been done to reveal Campylobacter's chicken and ruminant reservoirs across the world (Gormley et al., 2008; Wilson et al., 2008; Mullner et al., 2009; Rotariu et al., 2009; Sheppard et al., 2009). However, the focus of this research was on the risk of E. coli along the food supply chain. Several factors play a role in the likelihood of contamination of the food supply chain, which include factors related to the agent, some to the environment, and others are particular to the host. To develop a better appreciation of the complexity of the system, it is important to identify these factors that play a role in the survival and transmission of these pathogens in the food supply chain.
Conclusion
There was significant diversity among E. coli O157:H7 and food adulterant non-O157:H7 isolates along the food supply chain of animal origin and from patients; the transmission of these pathogens from the source to humans is not hierarchal. To maximize effectiveness of food safety policymaking, critical control interventions and proper sanitary and hygiene methods should be implemented at all levels.
Footnotes
Acknowledgments
This study would not have been possible without the Cornell University Institute of Biotechnology, EcMLST database curator Hans Steinsland, and Drs. Norval Strachan and Ovidiu Rotariu (University of Aberdeen) for their Excel VBAE program. The study was supported by grants from the Qatar Foundation (NPRP 07-292-3–070).
Disclosure Statement
No competing financial interests exist.