
Abstract
The ability to obtain bacterial genomes from the same host has allowed for comparative studies that help in the understanding of the molecular evolution of specific pathotypes. Avian pathogenic Escherichia coli (APEC) is a group of extraintestinal strains responsible for causing colibacillosis in birds. APEC is also suggested to possess a role as a zoonotic agent. Despite its importance, APEC pathogenesis still has several cryptic pathogenic processes that need to be better understood. In this work, a genome-wide survey of eight APEC strains for genes with evidence of recombination revealed that ∼14% of the homologous groups evaluated present signs of recombination. Enrichment analyses revealed that nine Gene Ontology (GO) terms were significantly more represented in recombinant genes. Among these GO terms, several were noted to be ATP-related categories. The search for positive selection in these APEC genomes revealed 32 groups of homologous genes with evidence of positive selection. Among these groups, we found several related to cell metabolism, as well as several uncharacterized genes, beyond the well-known virulence factors ompC, lamB, waaW, waaL, and fliC. A GO term enrichment test showed a prevalence of terms related to bacterial cell contact with the external environment (e.g., viral entry into host cell, detection of virus, pore complex, bacterial-type flagellum filament C, and porin activity). Finally, the genes with evidence of positive selection were retrieved from genomes of non-APEC strains and tested as were done for APEC strains. The result revealed that none of the groups of genes presented evidence of positive selection, confirming that the analysis was effective in inferring positive selection for APEC and not for E. coli in general, which means that the study of the genes with evidence of positive selection identified in this study can contribute for the better understanding of APEC pathogenesis processes.
Introduction
C
Positive selection can be detected by the analysis of molecular data using sophisticated statistical models based on the frequency of synonymous (dS) and nonsynonymous (dN) substitutions in homologous codons. The ratio of nonsynonymous substitutions/synonymous substitutions (ω = dN/dS) is commonly calculated to indicate what type of selection pressure is acting on a particular protein site. Thus, values of ω close to one suggests the occurrence of neutral evolution, ω <1 indicates purifying selection, and ω >1 indicates positive selection (Yang and Nielsen, 2002). When a nonsynonymous mutation occurs and results in a phenotypic change with a decrease in fitness for individuals carrying the new allele, the phenotype will be under purifying selection pressure, and the alleles will quickly be removed from populations (Ng and Henikoff, 2001). Nevertheless, some positions in certain genes can present a significantly higher than expected frequency of nonsynonymous mutations, indicating selection for the emergence of new alleles in place of the old gene copy. In pathogenic bacterial strains, enhanced fitness is frequently related to virulence- and niche-associated factors because these factors ensure bacterial survival and should be fixed in the population. An analysis of the substitution rates of nonsynonymous and synonymous mutations can reveal the action of positive selection on specific genes, with many of these genes being frequently involved in the parasite-host relationship (Yang and Bielawski, 2000; Vandamme, 2003; Aguileta et al., 2009, 2010).
Escherichia coli typically colonizes the gastrointestinal tract of birds and mammals in a commensal relationship. However, some strains have acquired specific virulence factors that enable adaptation to new niches, allowing these strains to cause different diseases (Russo and Johnson, 2000; Kaper et al., 2004). These features are very diverse and are associated with a many characteristics, such as adhesion (Verma et al., 2016), invasiveness (Pilatti et al., 2016), and improved fitness (de Paiva et al., 2015). Avian pathogenic E. coli (APEC) causes colibacillosis in birds, a disease that may be present as a localized or systemic infection, resulting in loss of production, decrease in egg production, carcasses condemnations, increased spending on treatment cost, and mortality, all factors that are responsible for severe losses in the multibillion dollar poultry production industry worldwide (Barnes et al., 2003). A subset of APEC presents significant similarities with E. coli isolated from cases of bacteremia and urinary tract infections in humans (Maluta et al., 2014), supporting a possible role of APEC as a zoonotic agent, probably as a foodborne pathogen.
Considering the importance of this pathotype, a comparative genomic study may help highlight possible adaptation processes that enable these strains to recognize and successfully survive in their host. Indeed, the comparison of genomic data from different strains within the same species allows an investigation of their evolutionary process, as well as phenotype modeling, because the similarities are sufficiently close to identify specific changes, and the differences are sufficiently minor to enable the detection of a large number of homologous genes (Petersen et al., 2007).
The study of genes under positive selection has previously been performed with E. coli strains. Using a branch-site model, 29 positively selected genes were identified in a uropathogenic E. coli (UPEC) strain (Chen et al., 2006), and 23 genes with evidence of positive selection were detected in E. coli and Shigella flexneri strains (Chen et al., 2006; Petersen et al., 2007). However, despite the huge importance of APECs for bird health, up to now there is not a comprehensive study searching and contextualizing the set of genes evolving with signs of recombination or positive selection in this pathotype.
Therefore, the goal of the present study is to perform a genome-wide search in APEC genomes present in the GenBank database to identify genes with evidence of recombination or evolving under positive selection. Knowledge of these genes can contribute to better understanding of APEC pathogenicity. Furthermore, these genes represent good candidates for future studies that would seek to analyze individual gene contributions to biological processes related to APEC pathogenesis.
Materials and Methods
Data acquisition and homology inference
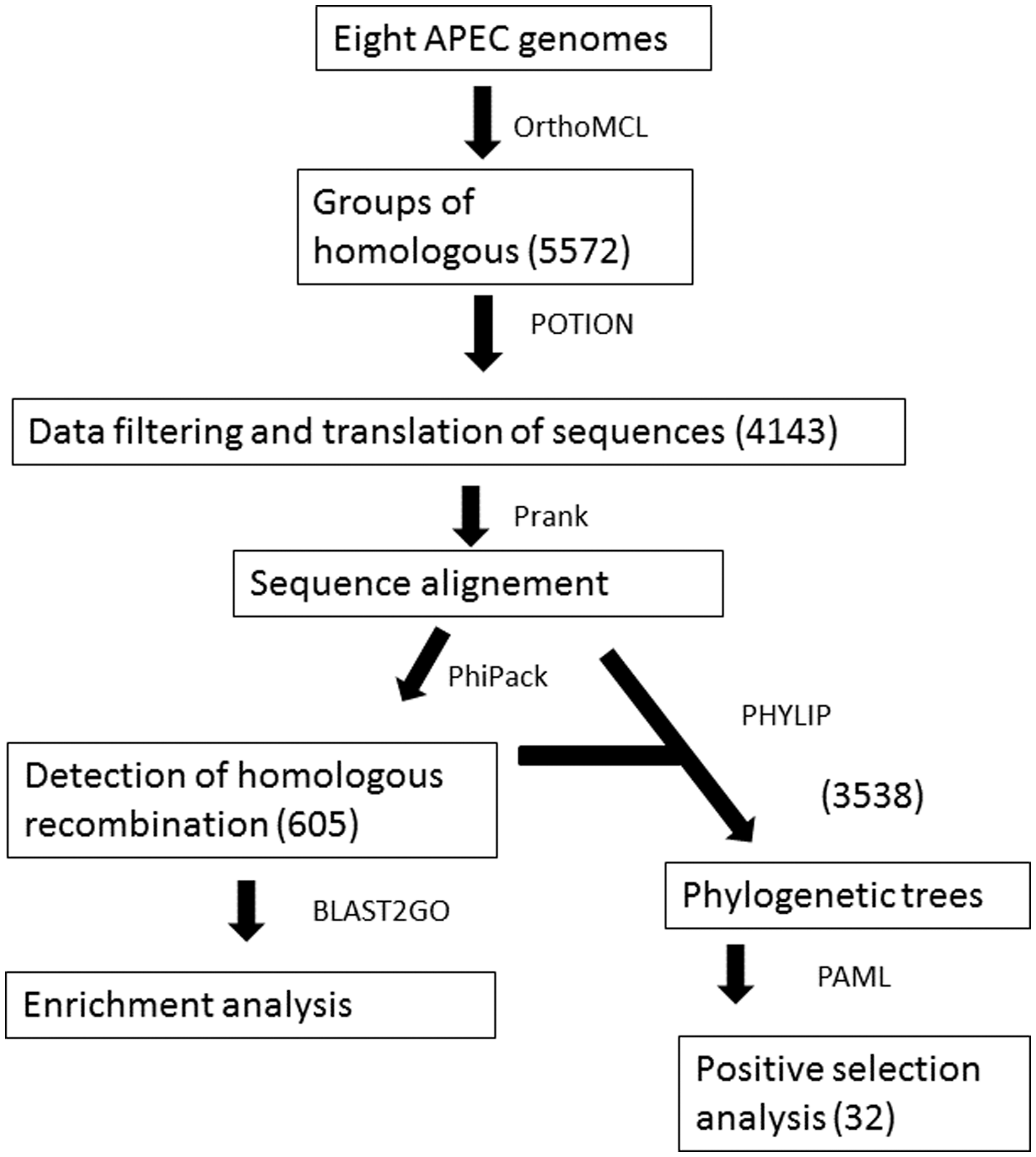
All APEC genome and plasmid sequences were obtained from the GenBank database. The genome set analyzed included eight APEC genome sequences, comprising two complete genomes and six draft assemblies (Table 1).
The groups of homologous genes were determined by OrthoMCL software (Li et al., 2003) using default parameters. We filtered out any sequences with no start codon and/or no stop codon (based on the NCBI bacterial codon table 11), sequences with ambiguous nucleotides and nonmultiples of three.
To verify whether the genes inferred to be under positive selection in APEC genomes are also under positive selection in other E. coli genomes that occupy other ecological niches, we selected E. coli strains belonging to two groups: one of them composed of only nonpathogenic strains from different origins, while the other was composed of human pathogenic strains (Supplementary Table S1; Supplementary Data are available online at
Experimental approach to detect genes under positive selection.
Data filtering and sequence alignment
We used a preliminary version of POTION to perform all the analysis from now on, except where explicitly stated otherwise (Hongo et al., 2015). It is well known that APEC represents a very diverse pathotype, with considerable variation in genome length and gene content (Table 1) (Maturana et al., 2011; Dziva et al., 2013). Therefore, analyzing only the 1:1 orthologs present in all eight genomes would result in the loss of valuable information. Furthermore, six of the eight genomes studied are draft versions that contain plasmid genes together with their assembled contigs. For these reasons, we chose to analyze the 1:1 orthologs present in at least four genomes, which is also a reasonable number from which to infer positive selection.
We also filtered out sequences from genomes with evidence of paralogy in any lineage, that is, duplicated sequences, leaving only putative 1:1 orthologs in the final set of sequences within each group. Finally, all groups with fewer than four sequences were excluded from further analyses. The groups remaining after all filtering steps are referred to hereafter as valid groups of homologs.
All the valid groups of homolog genes from the previous step were translated using the NCBI bacterial codon table 11, and the protein sequences were aligned using PRANK with default parameters (Löytynoja and Goldman, 2010). POTION uses an internal subroutine to generate protein-guided codon alignment files, which were used in downstream analyses.
Detection of homologous recombination and enrichment analysis
Homologous recombination was inferred from the codon alignment files for each valid group of homologs with PhiPack software, using all three recombination tests (Phi, NSS, and MaxChi2) (Bruen et al., 2006). All groups of homologous genes with a q-value smaller than 0.05, for at least two of the three tests, were considered as recombinants and were removed from the search for positive selection.
The groups with evidence of recombination were surveyed for possible enrichment in the context of Gene Ontology (GO) terms, using Blast2GO (Conesa et al., 2005). First, we annotated the longest sequence of each valid group of homologous using Blast2GO with default parameters. Afterward, the groups with evidence of recombination were surveyed for enrichment of specific GO terms (Fisher's Exact test, false discovery rate [FDR], and Benjamini–Hochberg correction). We used a FDR of 0.05 to control the inflation of Type 1 errors due to the multiple hypothesis testing scenarios.
Detection of genes under positive selection
The individual, multiple alignment files of protein sequences from all valid groups of homologs without evidence of recombination were used to reconstruct phylogenetic trees using proml from the PHYLIP package with 1000 bootstraps (Retief, 2000). The resulting phylogenetic trees and the multiple alignment files of codons previously obtained were utilized as input to the codeml from PAML 4.3 to detect possible groups of homologous genes with evidence of positive selection (Yang, 2007). We evaluated two popular nested codon evolution models (M1a × M2 and M7 × M8) to search for specific sites with evidence of positive selection in each alignment of a valid group. The search for positive selection in codeml was performed by comparing the log-likelihood values of codon models that do not allow sites with positive selection (M1a and M7) with the values of the more general coupled models that also do allow for site classes with the occurrence of positive selection (M2 and M8, respectively). The p-values were calculated as 2Δℓ (twice the difference in likelihood of the two nested models evaluated) based on the χ 2 distribution with two degrees of freedom. We used an FDR value of 0.1 to control the occurrence of false positive cases of positive selection due to the multiple testing scenarios.
Positive selection test was also performed on non-APEC strains. For that, the same genes detected in positive selection test performed on APEC strains were retrieved from the non-APEC genomes and grouped according to the homology. The same procedures for positive selection detection as described for APEC strains were performed separated for pathogenic and nonpathogenic strains.
Results
We obtained a total of eight APEC genomes and two plasmid sequences annotated from GenBank (Table 1) and identified 5572 homologous groups after OrthoMCL analysis. The genes likely to inflate false-positive selection detection, such as gene fragments, ambiguous coding sequences (CDS) or possible pseudogenes, and assembly errors, were then removed using the filtering procedures (“Materials and Methods” section). We also filtered out any genes with evidence of genomic duplication because orthologous and paralogous genes are likely to evolve under distinct selective pressures (Koonin, 2005). After the cleaning procedures, we obtained 4143 putative 1:1 orthologous groups in at least four APEC genomes. These groups, referred to as valid groups of homologs, were then subject to recombination and positive selection tests.
Recombination analysis determined that ∼14% (605) of the valid homologous groups showed significant evidence of homologous recombination. We performed an enrichment analysis searching for GO categories overrepresented in the list of genes with evidence of recombination (test set) compared with the total set of valid homologous groups (background set), to eventually gain a deeper insight about which gene categories may be target of recombination process in APECs. We found nine GO terms from the molecular function and biological process categories enriched in these genes. Enriched GO categories included ATP-dependent DNA helicase activity, ATP binding, DNA repair, tryptophan biosynthetic process, cofactor binding, metal ion transmembrane transporter activity, protein kinase activity, GTP binding, and ligase activity forming carbon-nitrogen bonds (Table 2).
C, cellular component; F, molecular function; P, biological process.
FDR, false discovery rate.
The 3538 valid groups of homologs with no evidence of recombination were tested for positive selection using the likelihood ratio test (LRT) for comparing two sets of popular nested codon evolution models implemented in codeml (M1a × M2 and M7 × M8). After multiple test correction considering a q-value smaller than 0.1, 69 genes with significant evidence of positive selection in at least one test remained. A total of 32 genes were detected by both models and were considered to be under positive selection for the APEC strains analyzed (Table 3). We performed enrichment analysis of these genes in the same way that was described for genes with homologous recombination, but in this case, the genes under positive selection were counterpoised against all valid group of homologous. In this analysis, 19 different GO classes were enriched (Table 2). Among these classes, the first three with smaller p-values and with two genes (viral entry into the host cell and detection of virus and pore complex) could be associated with bacterial defense. Furthermore, genes well known to be related with bacterial host interaction/contact were also detected (fliC, waaW, waaL, ompC). This preliminary analysis was able to show that several of the genes under positive selection can be viewed as important examples of the host-parasite defense/attack system, illustrating the premise that genes under positive selection are frequently related to virulence- and niche-associated factors.
The p-value and q-value have a cutoff value smaller than 0.05 and 0.1, respectively.
D represents the results of the likelihood ratio test for the set of models selected.
To confirm whether the positive selection results obtained for APEC strains were valid for E. coli in general, we separated two groups of E. coli strains, nonpathogenic and human pathogenic strains (Supplementary Table S1), retrieved from their genome the gene homologues to those 32 detected on APEC positive selection analysis and performed the same tests described above for APEC. No genes found to be under positive selection in APEC genomes were under positive selection in non-APEC genomes.
Discussion
Positive selection and recombination are important evolutionary forces involved in the phenotype-modeling process. The changes resulting from these two processes are responsible for genetic variations that may increase intraspecies genetic diversity and provide variability for the evolutionary process to act on (Xu et al., 2011). Recombination is an important force in bacterial pathogen adaptation, for several reasons. This phenomenon occurs at higher rates than mutation and, consequently, is a major force to introduce variability in a genome.
In this study, the enrichment analysis of the groups with evidence of recombination indicated that most genes were placed in ATP binding, ATP-dependent DNA helicase activity, GTP binding, DNA repair, and cofactor binding classes. These enriched classes include proteins with diverse biological functions, such as transport activity, sensory regulators, cell division, metabolism, chaperones, and resistance to antimicrobials. The high number of GO terms related to ATP-interacting proteins can lead us to think that this could be the result of a recombination bias for this type of protein because of the ATP needs of RecA (recombinase A) (Goodman, 2014).
The comparative, evolutionary genomic study of different strains through the detection of genes that are under positive selection allows for the identification of factors that may provide an adaptive advantage to these strains. In general, it has been shown that a large proportion of genes under positive selection have a close relationship with the organism lifestyle, in the specific case of pathogenic strains, with parasitism-related genes (Chen et al., 2006; Petersen et al., 2007; Xu et al., 2011). Thus, greater knowledge of genes under positive selection may help further understand the pathogenicity processes and, consequently, aid the development of vaccines and drugs (Polley et al., 2003).
As a pathogen of major importance, several pathotypes of E. coli have been previously surveyed for positive selection. In a search for positive selection in specific lineages of phylogenetic tree (branch-site tests), 29 genes under positive selection were identified in the UTI89 strain. Several of the detected genes were involved in cell surface structure, nutrient acquisition, DNA metabolism, and urinary tract infection processes. Another comparative genome study identified 23 genes with evidence of positive selection in E. coli and S. flexneri strains (Petersen et al., 2007). Most of the gene products were predicted to be located on the cell surface and involved in interactions with the host, phage, or other bacteria, suggesting a coevolutionary context (Chen et al., 2006; Petersen et al., 2007).
When we compare the genes that were identified to be under positive selection with genes identified in the E. coli-related studies described above, we note that the ompC product is commonly identified in all studies (Chen et al., 2006; Petersen et al., 2007). OmpC is a trimeric porin with a B-barrel domain localized on the outer membrane that allows permeability for the transport of ions and hydrophilic solutes across membranes (Nikaido, 1994; Schirmer, 1998). OmpC is one of the most abundant proteins expressed in E. coli and has been demonstrated to be expressed at significantly higher levels during urinary tract infections. High expression of OmpC in vivo may be an advantage in environments, where small molecular toxins are plentiful because the pore formed by OmpC also serves as an exit path for toxins (Nikaido, 1994). Another important feature of this protein is that the pore formed by OmpC also functions as a receptor for various phages (Yu et al., 2000).
The beta barrel porin LamB, found to be under positive selection in this study and in the study conducted by Petersen et al. (2007), is a maltose outer membrane porin, the main function of which is the diffusion of maltose (Wang et al., 1997). The gene name refers to the Lambda phage because this porin also operates as a receptor for this phage (Randall-Hazelbauer and Schwartz, 1973). The four sites found to be under positive selection were residues 408, 409, 410, and 411 of the 446-amino acid LamB protein, corresponding to the loop 9 region involved in phage binding (Newton et al., 1996). These results are similar to those obtained by Petersen et al. (2007), suggesting that LamB positive selection may be the result of selection to avert phage binding.
Two other genes under positive selection found in our study, waaL and waaW, are related to the biosynthesis of the outer membrane lipopolysaccharide (LPS). The gene waaW product is the enzyme UDP-galactose, (galactosyl) LPS alpha 1,2-galactosyltransferase, that acts during LPS biosynthesis. LPS consists of three parts as follows: lipid A, the core oligosaccharide (OS) and O-antigen (Raetz and Whitfield, 2002; Valvano, 2011). The gene waaL product is required to ligate the terminal sugar of lipid A to OS (Valvano, 2011); the absence of WaaL results in rough bacteria with no O-antigen polysaccharide (Mulford and Osborn, 1983; McGrath and Osborn, 1991). WaaL is a promising target for antivirulence factors due to O-antigen host–pathogen interactions and its localization in the periplasmic space (Ruan et al., 2012).
Pathogenic E. coli strains can be identified by O:H serotyping, which corresponds to the cell surface LPS O-antigen and the flagellar antigen H (Orskov et al., 1977). The fliC gene, observed to be under positive selection in our study, encodes the structural subunits of flagellin. The comparison of several homologous flagellin sequences revealed that the N- and C-terminal regions are well conserved among species. In contrast, the central region is rather variable and is responsible for the antigenic property of the filament, closely interacting with the host defense system (Wieler et al., 1997). Our positive selection analysis revealed that the sites with higher nonsynonymous substitution rates are in the central region of the protein sequence. As fliC positive selection can be the result of the intimate contact between the host and bacterial cells during infection, selection may function to avoid bacterial recognition by the host immune system.
The phage-shock-protein A (PspA) was first discovered during an E. coli filamentous phage infection due to its high concentrations (Brissette and Russel, 1990). The Psp system includes proteins PspA, B, C, D, E, F, and G and responds to several factors responsible for damage to membrane integrity, such as filamentous phage infection, medium downshifts, extreme temperatures, the mislocation of some envelope proteins, osmolarity, ethanol concentration, and the presence of proton ionophores, such as carbonylcyanide m-chlorophenylhydrazone (CCCP) (Model et al., 1997). This regulon is required to maintain the proton-motive force that protects the inner membrane, and this stress adaptation is important for bacterial growth under stress conditions and for pathogenicity (Darwin, 2005). The sensory proteins PspB and PspC positively regulate the expression of PspA through protein–protein interactions under stress conditions resulting in the release of the inhibitory complex PspA-PspF and in an increased expression of other Psp proteins (Jovanovic et al., 2006). PspA, which is under positive selection, can be considered an example of how selection acts on factors that enable bacterial survival under stress conditions.
After reviewing all genomic information currently available, we found that several genes have still not been characterized. In this positive selection study, some genes corresponding to uncharacterized protein functions were detected (yjgL, ymfN, ydiE, yicS, ydjO, A364_13802, and A364_15858). The identification of unknown genes with occurrence of positive selection may help in further understanding of the processes of APEC pathogenicity. Even though several of these gene activities remain cryptic, they constitute promising targets for study and, if their role in bacterial pathogenesis were demonstrated, they may constitute interesting targets for the development of attenuated vaccine strains.
Conclusions
In conclusion, the genes found to be under positive selection in APEC were not under positive selection in non-APEC E. coli. This result may indicate that, in our restricted analysis, the genes that are under positive selection in APEC appear to be specific of this pathotype and may represent an APEC response to environmental pressure.
Footnotes
Acknowledgments
The authors thank the Laboratório Multiusuário de Bioinformática of Embrapa Informática Agropecuária for the computational resources. This project was supported by grant numbers 2010/51421-8 (FAPESP), 2012/04931-6 (FAPESP), PNPD0146080 (CAPES), and 485279/2011-8 (CNPq).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.