
Abstract
In the detection of Shigella species using molecular biological methods, previously known genetic markers for Shigella species were not sufficient to discriminate between Shigella species and diarrheagenic Escherichia coli. The purposes of this study were to screen for genetic markers of the Shigella genus and four Shigella species through comparative genomics and develop a multiplex polymerase chain reaction (PCR) for the detection of shigellae and Shigella species. A total of seven genomic DNA sequences from Shigella species were subjected to comparative genomics for the screening of genetic markers of shigellae and each Shigella species. The primer sets were designed from the screened genetic markers and evaluated using PCR with genomic DNAs from Shigella and other bacterial strains in Enterobacteriaceae. A novel Shigella quintuplex PCR, designed for the detection of Shigella genus, S. dysenteriae, S. boydii, S. flexneri, and S. sonnei, was developed from the evaluated primer sets, and its performance was demonstrated with specifically amplified results from each Shigella species. This Shigella multiplex PCR is the first to be reported with novel genetic markers developed through comparative genomics and may be a useful tool for the accurate detection of the Shigella genus and species from closely related bacteria in clinical microbiology and food safety.
Introduction
S
The outbreaks of shigellosis are primarily caused by contact with infected individuals and have been associated with the consumption of contaminated food or water (Warren et al., 2006; Lin et al., 2010). The phenotypic and genotypic characteristics of Shigella species are reported as being too similar for distinguishing from diarrheagenic Escherichia coli (enterohemorrhagic E. coli, enteropathogenic E. coli, enterotoxigenic E. coli, enteroinvasive E. coli [EIEC], and enteroaggregative E. coli), reflecting the evolutionary and taxonomical history of the Shigella and Escherichia genera, particularly EIEC, which is a major cause of dysentery and is regarded as the evolutionary ancestor of the Shigella (Lan et al., 2004; Yang et al., 2005). The generally known phenotypic differences of Shigella from E. coli are its lack of lactose fermentation and absence of motility.
For the understanding of epidemiological and pathological features of Shigella, genotype-based comparisons have been performed, including the sequence analysis of virulence factor genes, housekeeping genes, and whole genomes (Fukiya et al., 2004; Lan et al., 2004; Yang et al., 2005). In addition, microarray-based comparative genomics have been applied to differentiate the gene contents between Shigella and E. coli for a better understanding of the evolutionary history, diversity, and pathogenesis of Shigella species (Fukiya et al., 2004; Peng et al., 2006).
For reliable identification of Shigella spp. and each of the four Shigella species from closely related genera, polymerase chain reaction (PCR)-based methods have been reported using target genes of virulence factors in plasmid DNA: invasion plasmid antigen H (ipaH) (Aranda et al., 2004; Binet et al., 2014), ial (Sethabutr et al., 1993; Thong et al., 2005), virA (Villalobo and Torres, 1998), the she pathogenicity island (Farfan et al., 2010), and tuf (Maheux et al., 2011). Although PCR is a reliable method for the identification of bacteria, and despite efforts on the specific detection of Shigella spp., shigellae is still regarded as indistinguishable from EIEC (or diarrheagenic E. coli) according to PCR or other molecular methods (Venkatesan et al., 1989; Villalobo and Torres, 1998; Warren et al., 2006). Furthermore, each species of Shigella is regarded as indistinguishable from other Shigella species according to PCR-based methods (Thong et al., 2005; Farfan et al., 2010; Pavlovic et al., 2011). Recently, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry was attempted to differentiate between Shigella species and E. coli, but reported some discrepant results at the genus/species levels (Khot and Fisher, 2013).
The aims of our study are to screen genetic markers of shigellae and each of Shigella species and to suggest a method of distinguishing the four Shigella species with specificity. The genome sequences of Shigella species were analyzed using comparative genomics to screen for specific gene(s). These specific primer sets, designed from the screened genes, of shigellae and each four Shigella species were evaluated with various strains of Enterobacteriaceae and other pathogenic bacteria, including Shigella and diarrheagenic E. coli. This Shigella multiplex PCR was first suggested as an accurate means of distinguishing shigellae and four Shigella species from each other, from E. coli, and from closely related bacteria for applications in clinical microbiology and food safety.
Materials and Methods
Bacterial strains and genomic DNA extraction
Strains of Shigella and diarrheagenic Escherichia coli were collected from the American Type Culture Collection (ATCC) and the National Culture Collection for Pathogens (Korea) (Table 1). The Shigella strains were inoculated in a nutrient broth medium and cultured at 37°C with vigorous shaking. Non-Shigella-type strains, including foodborne pathogens and Enterobacteriaceae, were collected from the ATCC. Bacterial genomic DNA was extracted using the G-spin Total DNA Extraction Mini Kit (Intron) and genomic DNA purified to a spectrophotometric ratio (A260/A280) of 1.8–2 was used.
ATCC, American Type Culture Collection; NCCP, National Culture Collection for Pathogens.
Comparative genomics for screening genetic marker of each Shigella species
The genome sequences and sources of Shigella strains are shown in Table 2. One representative genome sequence of each target Shigella species was used for species-specific gene screening: Shigella sonnei Ss046, Shigella boydii Sb227, Shigella dysenteriae Sd197, and Shigella flexneri 2a str. 301. To screen specific genes, the coding region sequences (ffn file) of each Shigella species (target Shigella) were BLASTed against the database, which consisted of genomic DNA sequences (fna file) of the Shigella species, excluding the genome sequences of the particular target Shigella species, using the Basic Local Alignment Search Tool (BLAST) program (version 2.2.13) (Altschul et al., 1997; Begley et al., 2010; O'Flaherty and Klaenhammer, 2011). Based on the BLAST outputs, for each target Shigella species, we selected gene sequences that had low homology scores relative to the genomes of other Shigella species, re-BLASTed them against the nonredundant (nr) database in the NCBI, and screened the candidate genes for each specific Shigella species to design the primer sets.
The information in this table was updated in August 2012.
Completed genome sequences were obtained from the National Center for Biotechnology Information (NCBI,
Comparative genomics for screening genetic marker of shigellae
For screening shigellae-specific DNA fragments, the coding region sequences of Shigella flexneri 2a str. 301 were used as representative genome sequences of shigellae. The coding region sequences were divided into 500-base pair (bp) segments, overlapping every 400 bp (shifting 100 bp), and the entirety of the 500 bp segments was BLASTed against each representative genome sequence of Shigella boydii Sb227, Shigella sonnei Ss046, and Shigella dysenteriae Sd197 in order. From the output of the BLAST, DNA fragments that showed a high degree of homology with all other Shigella species and were expected to be present in all Shigella species were BLASTed against a database consisting of 29 E. coli genome sequences (list not shown). From the output, DNA fragments with low homology with the E. coli database were selected to screen for candidate shigellae-specific DNA fragments. Finally, selected DNA fragments were compared with the nr database and the genome database of microbes for design of the primer sets.
Primer construction and PCR conditions
Primer sets expected to be specific for each Shigella species and shigellae were designed from each screened candidate gene (or DNA fragment). These primer sets were evaluated with each of the genomic DNAs of Shigella and the other type strains shown in Table 1. PCR amplifications were carried out with 200 mM of each dNTP, 0.5 U of ExTaq DNA polymerase (TaKaRa Bio, Inc.), 1 × ExTaq buffer (Mg2+ plus), 25 ng of template DNA, and the adjusted concentration of each primer in a final reaction volume of 25 μL. PCR amplification was performed in a thermocycler (PC-808; ASTEC) with an initial denaturation at 94°C for 5 min, followed by 25 cycles of 94°C for 30 s, 63°C for 30 s, and 72°C for 30 s, finishing with a final extension at 72°C for 5 min. Amplified products were electrophoresed on a 2.5% agarose gel in 0.5 × Tris-acetate-ethylenediaminetetraaceticacid buffer, stained with ethidium bromide, visualized under UV irradiation, and photographed with a digital camera (Nikon; COOLPIX 4300).
Multiplex PCR of Shigella and construction of the internal amplification control
Multiplex PCR was performed using finally selected five primer sets. The mixture and conditions of multiplex PCR for one reaction were the same as in the single PCR except the primer concentrations (Table 3) and internal amplification control (IAC) template plasmid DNA (4 pg, ∼106 copies). The IAC was constructed based on the finally selected gene of Shigella sonnei Ss046 (NC_007384: 1665285-1666367) included in the multiplex PCR. A primer set was designed as SS1665285 IAC-F (5′-GCAGCACTCTTTGATGCCGGG CTGATGCCGTAGTCGTCACT-3′) and SS1665285 IAC-R (5′-CCCGTTCGGTCCTCTCCCAAAACGGGCCCGGAGCTAAAGTT-3′). These primers were flanked with the primer sequence of SS1665285-F421 and SS1665285-R809 at the 5′ end, enabling the amplification of a 100- bp PCR product with constructed IAC. This primer set for IAC construction was amplified with genomic DNA from S. sonnei using PCR for subsequent cloning in the pGEM-T easy vector (Promega).
Reference sequence number of chromosomes at NCBI and position of gene.
PCR, polymerase chain reaction.
Results
Genetic marker screening and primer design for each Shigella species and shigellae
Among the coding region sequences of each Shigella species, ∼110–230 genes with a low homology output (matching under <25 [or 30] bp of nucleotide) were selected using the BLAST program for comparisons of the Shigella genome database to first screen for Shigella species-specific genes. These selected genes were then BLASTed to the nr database of NCBI; from each Shigella species, ∼10–40 genes with a relatively low homologous output were selected to eliminate relatively highly homologous genes that were closely related to other bacteria. The selected genes were BLASTed against the NCBI microbial genome database. Finally, from each Shigella species, between two and four genes that were expected to be species specific were selected based on their low homology within Shigella genus and with other bacteria.
In the screening of specific DNA fragments for shigellae, the coding region sequences of Shigella flexneri 2a str. 301 were divided into more than 35,000 of 500 bp-sized segments and BLASTed against each genome sequence of S. sonnei, S. boydii, and S. dysenteriae. More than 29,000 fragments that were expected to be core genes of shigellae were screened from the output of the BLAST results and were BLASTed against a database that consisted of 29 E. coli genome sequences to avoid the sequences of E. coli overlapping with shigellae and screen for low homology DNA segments of shigellae. Finally, 50 DNA segments with the lowest degrees of homology with the E. coli genome database were selected and confirmed with the microbial genome database of NCBI.
Specific primer sets were designed from the selected specific candidates considering the BLAST output of the microbial genome database, PCR product size, specificity, % GC content, and conserved regions within shigellae and other closely related bacteria.
The specificity of designed primer sets of shigellae and each Shigella species
A total of 15 primer sets were designed and their specificities to shigellae and each Shigella species were evaluated using conventional PCR, including a total of 36 various bacterial strains of Shigella and E. coli, as well as other pathogenic bacteria and Enterobacteriaceae (Table 1). Although these primer sets were designed from the genes expected to be specific based on the comparative genomics of Shigella, one-fourth of the primer sets failed to amplify specific DNA fragments of the expected sizes. After the evaluation of each primer set, one of each specific primer set for shigellae, S. flexneri, S. sonnei, S. dysenteriae, and S. boydii were selected considering the melting temperature and the intensity of amplified PCR product. These selected primer sets produced specific bands only with target Shigella species or shigellae at the expected sizes, and there were no amplified bands when the primers were used with diarrheagenic E. coli or other bacterial strains examined in this study (data not shown).
Design of the multiplex PCR for shigellae and its specificity
Using the five primer sets selected from single PCR confirmation based on specificity, a multiplex PCR was developed for identification of shigellae, S. flexneri, S. dysenteriae, S. boydii, and S. sonnei (Table 3), including the source of the gene, primer concentrations, and sequences. The concentration of each primer set was adjusted based on the intensity of the amplified bands through repeated multiplex PCR performances. Constructed plasmids, including IAC, which were designed to amplify a 100 bp PCR product with the primer set of SS1665285 F421-R809 to confirm the reaction, were also contained in this multiplex PCR. This multiplex PCR was evaluated with various genomic DNAs of bacterial type strains (Fig. 1). This multiplex PCR could amplify a total of six bands, including IAC (Fig. 1, Lane 1), and each Shigella strain showed a shigellae-specific PCR product (159 bp), as well as a Shigella species-specific PCR product (S. flexneri, 132 bp; S. dysenteriae, 190 bp; S. boydii, 240 bp; S. sonnei, 389 bp) (Fig. 1, Lane 2–7). Neither diarrheagenic E. coli, not even EIEC, nor any other pathogenic bacteria produced PCR products, demonstrating the accuracy and specificity of this multiplex PCR. The multiplex PCR results illustrated that these primer sets were clearly able to discriminate between specific Shigella species and between shigellae and other closely related bacteria. In addition, the limit of detection (LOD) and multidetection ability of the Shigella multiplex PCR using genomic DNA combinations from Shigella species were confirmed. Also, the applications of Shigella multiplex PCR in food matrices (lettuce and beef) were evaluated. The LODs were between 5 × 104 and 5 × 103 copies of Shigella genomic DNA in a 25-cycle PCR and between 5 × 103 and 5 × 102 copies in a 30-cycle PCR (Supplementary Figs. S1 and S2; Supplementary Data are available online at
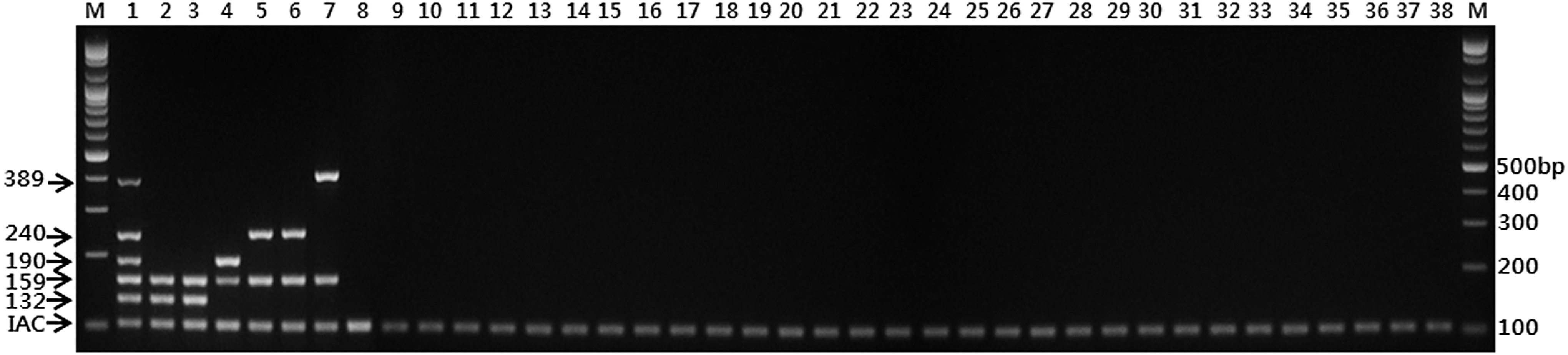
Results of Shigella multiplex polymerase chain reaction with bacterial strains of Enterobacteriaceae and pathogenic bacteria. M: 100 bp DNA ladder; lane 1: DNAs including four Shigella species (S. flexneri ATCC 12022, S. dysenteriae ATCC 13313, S. boydii ATCC 8700, S. sonnei ATCC 25931); lane 2: S. flexneri ATCC 12022; lane 3: S. flexneri 2a strain 2457T ATCC 700930; lane 4: S. dysenteriae ATCC 13313; lane 5: S. boydii ATCC 8700; lane 6: S. boydii ATCC 9905; lane 7: S. sonnei ATCC 25931; lane 8: enteroinvasive Escherichia coli ATCC 43893; lane 9: enteroaggregative E. coli NCCP 14039; lane 10: enteropathogenic E. coli NCCP 14038; lane 11: enterotoxigenic E. coli NCCP 14037; lane 12: E. coli O157:H7 ATCC 43890; lane 13: E. coli O157:H7 ATCC 43894; lane 14: E. coli ATCC 35150; lane 15: E. coli ATCC 11775; lane 16: Salmonella enterica serovar Typhimurium ATCC 19585; lane 17: Salmonella serovar Typhi ATCC 33459; lane 18: Salmonella serovar Enteritidis ATCC 4931; lane 19: Salmonella serovar Gallinarum ATCC 9184; lane 20: Salmonella serovar Pullorum ATCC 9120; lane 21: Yersinia enterocolitica ATCC 29913; lane 22: Enterobacter aerogenes ATCC 13048; lane 23: Enterobacter cloacae ATCC 13047; lane 24: Cronobacter sakazakii ATCC 29544; lane 25: Proteus vulgaris ATCC 29905; lane 26: Citrobacter freundii ATCC 8090; lane 27: Rahnella aquatilis ATCC 15552; lane 28: Bacillus cereus ATCC 14579; lane 29: Listeria monocytogenes ATCC 19113; lane 30: L. seeligeri ATCC 35967; lane 31: L. innocua ATCC 33090; lane 32: Vibrio parahaemolyticus ATCC 27969; lane 33: V. vulnificus ATCC 33815; lane 34: V. cholerae NAG KCDC 13589; lane 35: Staphylococcus aureus ATCC 29737; lane 36: S. epidermidis ATCC 14990; lane 37: S. haemolyticus ATCC 29970; lane 38: no template.
Discussion
The Shigella genus was taxonomically distinguished as a different genus from E. coli in 1940s and is considered to be evolutionarily derived from EIEC (Peng et al., 2009). Due to its evolutionary history and similar genome content to E. coli, identification method using molecular biological techniques, including PCR for the accurate differentiation of shigellae from E. coli and between Shigella species, had not been developed. Particularly, EIEC are biochemically similar to shigellae, and some EIEC are also serologically similar to shigellae. Despite previous efforts to develop an identification method for each Shigella species using PCR, the limited number of target DNA sequences, especially virulence factor genes such as ipaH and virA, necessitated the need for additional reliable genetic markers that differentiate between Shigella species and E. coli (Aranda et al., 2004; Warren et al., 2006; Lin et al., 2010; Maheux et al., 2011). In addition, these virulence factor genes present in the plasmid of Shigella could lead to false-positive or false-negative results in PCR due to the horizontal transfer of plasmids to other genera in Enterobacteriaceae, which share close evolutionary histories, or the loss of the plasmid in particular circumstances. Moreover, along with the identification of shigellae from Enterobacteriaceae, more detailed and reliable diagnostics for Shigella at the species level is also of benefit in aiding in an understanding of their features and pathogenic characteristics in food safety and clinical microbiology, as well as reflecting their regional population differences (Kotloff et al., 1999).
As expected and reported from a comparative genomics microarray study, which characterized the different gene contents of each Shigella species for gene presence/absence levels (Fukiya et al., 2004; Peng et al., 2006), many candidate genes from our comparative genomics study were determined to be unique to an individual Shigella species, although their protein functions were not clearly known, and some of the candidate genes matched a couple of whole-genome shotgun sequences from E. coli in the draft genome database of NCBI. In contrast with the screening process for genetic markers in each Shigella species, certain other factors were considered in the screening process for genetic markers of generic shigellae. First, the core genes of shigellae. Candidate genes for a genetic marker of generic Shigella must be present in all Shigella species. To identify the core genes of shigellae, gene sequences of S. flexneri 2a str. 301, which has a relatively large genome size and gene number, were BLASTed against each genome sequence of the Shigella species in order in this study. Second, the ability of genetic marker to differentiate shigellae from E. coli. Although the core genes of shigellae were selected, we considered that most of the genes would be present in and similar to E. coli, in particular EIEC. To overcome the similarities of the Shigella and E. coli genomes, the coding region sequences of S. flexneri were divided into 500 bp segments, shifting 100 bp at the first stage for a more detailed analysis of sequences, and a database of 29 E. coli genome sequences was compared to remove the genes that overlapped with those of E. coli.
In our previous studies, “in silico” comparative genomics were successfully applied and shown to be applicable to PCR and microarray for genotyping and detection of Salmonella and other pathogenic bacteria (Kim et al., 2006a, b, 2008). Despite our efforts in this study to screen for shigellae-specific sequences, the core genes shared between shigellae and E. coli were so similar that it was difficult to determine shigellae-specific sequences. The ultimately selected sequences for a shigellae-specific primer set in the multiplex PCR had some degree of matching with the whole-genome shotgun sequence of one E. coli in a draft genome database in NCBI (not matched with the nr and completed genome database in NCBI; data not shown). However, this weak point of the shigellae-specific primer set could be supplemented in our multiplex PCR by using the other four primer sets specific to each Shigella species, providing results at the Shigella species level for more accurate identification. In addition, potential application of this Shigella multiplex PCR for food industry and clinical microbiology field was evaluated by the estimation of LODs with Shigella genomic DNA and artificially Shigella-spiked food as shown in Supplementary Figures S1 and S2 and Supplementary Table S1. For the Shigella diagnostics in food sample, the LOD of Shigella multiplex PCR revealed not able to detect low number (100–200) of cells, which can cause shigellosis when ingested through food. However, we anticipate clinical application of this assay with stool of shigellosis patient, which contains relatively high number of Shigella (up to a million per gram).
Conclusion
We demonstrated the success of comparative genomics in screening for genetic markers of generic Shigella and each of four Shigella species. After evaluation of the primer sets, designed from screened genetic marker, with Shigella, E. coli and other bacterial strains, a multiplex PCR was developed for the identification of Shigella genus/species and was confirmed with Shigella and E. coli strains, demonstrating its specificity. Unique characteristics of the Shigella multiplex PCR include novel genetic markers and diagnostic ability, which both result in simultaneous detailed identification at the Shigella genus and species level. Also, the performance of Shigella multiplex PCR demonstrated the ability to distinguish shigellae and each Shigella species from diarrheagenic E. coli. Despite the fact that more improvement of these genetic markers developed in this Shigella multiplex PCR will be needed to maximize their reliable diagnostic ability for further applications in practical fields, we suggest that the Shigella multiplex PCR revealed sufficient specificity and applicable ability as a useful diagnostic tool for Shigella genus and species.
Footnotes
Acknowledgment
We thank Dr. Hyo-Sun Kwak, Korea National Institute of Health, for providing Shigella strains.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.