
Abstract
Background:
Vibrio parahaemolyticus is an emerging foodborne pathogen in the Mediterranean, usually associated with shellfish consumption. The increase in the number of outbreaks in Europe is primarily associated with the global warming of the ocean that has a great impact on the spread and genetic selection of waterborne pathogens. The primary role of Italy in Europe's mollusk production, together with the fact that cases of infections with V. parahaemolyticus are not always notified to the European community, highlighted the necessity of acquiring new information about the epidemiological involvement of shellfish products.
Objective:
The aim of the study was to provide useful insights into the first steps of the Risk Assessment associated with V. parahaemolyticus through the molecular characterization of isolates from commercialized mollusks.
Materials and Methods:
A total of 102 strains identified as V. parahaemolyticus were investigated as part of a larger sampling (1-year survey) from several shellfish species collected from the Venice lagoon and the North Adriatic sea. All strains were characterized by multilocus sequence typing and tested for the presence of virulence genes (trh and tdh). The study of sampling/environmental factors and epidemiological analyses was performed to describe the behaviors of the different genetic populations.
Results:
The population structure analysis highlighted three genetic clusters that could be subject to temperature selection during cold (≤15°C) and warm (>16°C) seasons. Moreover, other factors, such as molluscan species (clams/mussels), probably played a role in the distribution of genetic clusters. Although few strains carried the virulence factors (n = 6 trh +), epidemiological links with clinical isolates and a local dissemination of some sequence types were underlined.
Conclusion:
This work provides a useful background on the genotype spread as a first step in the Hazard Identification in light of future climate changes.
Introduction
V
During the last two decades, V. parahaemolyticus infections and outbreaks associated with the consumption of raw or undercooked shellfish have increased throughout the world (Haendiges et al., 2015), recognizing the bacterium as a main pathogen of seafood-borne diseases in many countries, especially in Asia, the United States, and Africa (Rojas et al., 2011).
Even though infections due to V. parahaemolyticus have rarely been reported in Europe (FAO/WHO, 2011), the recent isolation of potentially pathogenic strains on this continent demonstrates that the health risk associated with V. parahaemolyticus infection is increasing.
Strains with potential pandemic markers and virulence genes were isolated from clinical sources in central Italy (Ottaviani et al., 2008) and in environmental strains in the northern Adriatic Sea and Southern Italy (Di Pinto et al., 2008; Caburlotto et al., 2010). Moreover, other cases of V. parahaemolyticus gastroenteritis in Italy were associated to the O1:KUT serotype (Ottaviani et al., 2009). The spread of new pathogenic clones and the increase of outbreaks registered in Europe have been hypothesized to be related to a plethora of factors mainly associated with climate change (Martinez-Urtaza et al., 2008). The rise in water temperature and the extreme meteorological precipitations associated with heat waves opened a new global scenario on the risks related to this pathogen (Baker-Austin et al., 2017).
Italy produces 150,000 tons of marine mollusks per year, being the third largest producer in Europe. Moreover, the Veneto region plays a significant role at the national level, as it is an important producer of mussels and clams (Robert et al., 2013). These lines of evidence, together with the considerable public health implications of this type of infection, suggest that monitoring V. parahaemolyticus in shellfish is crucial.
Currently, the Regulation EC (2073/2005) is not compulsory for the identification of potentially pathogenic Vibrio spp. However, the need to develop reliable methods and new criteria for the identification of V. parahaemolyticus has already been mentioned in the European legislation. This should prompt official veterinary and health authorities to pay more attention to the epidemiological roles of environmental microorganisms in local foodborne diseases and to increase the microbial surveillance of pathogenic V. parahaemolyticus strains (Ottaviani et al., 2008). In particular, the molecular typing of strains circulating in the environment can provide a more complete epidemiological picture. In fact, very little data are currently available about the prevalent genotypes of Italian strains.
Multilocus sequence typing (MLST) was suggested as a fast and consistent method to produce reliable typing of organisms (EFSA, 2013), producing sequences suitable for additional comparative analyses.
In this study, MLST has been successfully applied to characterize 102 strains isolated from mollusks in the North Adriatic Sea. The MLST method produced a local-strain database to study their phylogenetic relationships, genetic structure, and genetic correlation with strains isolated in other countries and from different sources. It is clear that datasets containing information useful for the microbial source tracking of Vibrio species, namely in Europe, must be made available promptly (Baker-Austin et al., 2017). This work outlines information on the variability of the strains, their potential virulence, and the epidemiological links with worldwide strains. The combination of sampling/ecological information and molecular data made it possible to define the most important factors (e.g., temperature) affecting the genetic population behaviors.
Materials and Methods
Sampling, isolation, and identification of V. parahaemolyticus strains
A total of 130 samples of mollusks were collected during 2011 during different seasons and in several areas of the Veneto region (North Adriatic Sea -Table 1, Fig. 1 and Supplementary Fig. S1; Supplementary Data are available online at
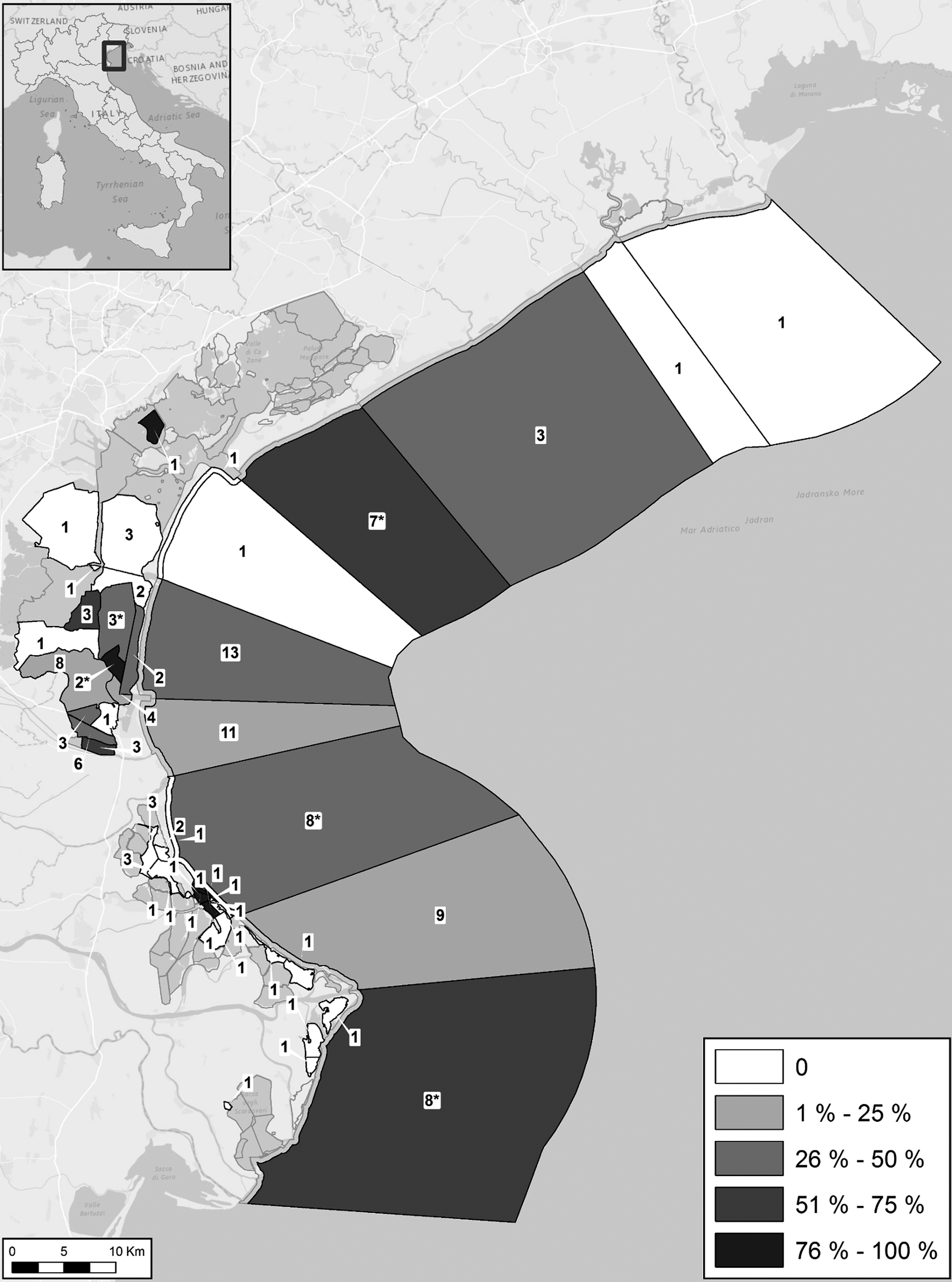
Map of the sampling area, showing the number of mollusks collected and the percentage of positive samples for Vibrio parahaemolyticus at each sampling point. The symbol (*) represents the samples positive for trh+ strains.
W°C, water temperature; A°C, air temperature; values were referred to both lagoon and sea sites.
The samples were processed following [ISO/TS] 21872-1:2007 using the selective media Thiosulfate-Citrate-Bile Salts-Sucrose Agar (Oxoid, Basingstoke, United Kingdom) and Chromagar Vibrio plates (CHROMagar Microbiology, Paris, France; see Supplementary Material and Methods), and first-level identification of V. parahaemolyticus was performed by classical biochemical tests (Alsina and Blanch, 1994).
Total DNA extractions from strains were performed as described by Martino et al. (2011). The strains were subjected to molecular confirmation by amplification of toxR and tlh genes with species-specific primers (Taniguchi et al., 1986; Brasher et al., 1998; Kim et al., 1999) and by multilocus sequence analysis (MLSA) identification using four housekeeping genes (gyrB, pyrH, recA, and atpA) developed by Rahman et al. (2014).
Molecular typing (MLST) and identification of virulence genes
All strains recognized as V. parahaemolyticus were further characterized using the MLST scheme developed by Gonzalez-Escalona et al. (2008). The primers used to amplify internal fragments of the seven housekeeping genes (gyrB, dnaE, dtdS, pntA, pyrC, recA, and tnaA) are described on the V. parahaemolyticus PubMLST website (
MLST data analyses, population structure, and effects of ecological/sampling factors
Chromatograms and sequences obtained for the 102 strains of V. parahaemolyticus were processed following the pipeline described by Martino et al. (2014). The V. parahaemolyticus MLST database was used to derive both the ID numbers of each allele present in our dataset and the sequence types (STs) derived from the allelic profiles. New alleles were sent to the curator of the V. parahaemolyticus PubMLST website to be included in the database. The concatenated sequences were aligned and used to construct a maximum likelihood phylogenetic tree using MEGA v.6 (Tamura et al., 2011).
Structure software (Falush et al., 2003) was used to investigate the population structure (distinct clusters and/or mixed genomic profiles), accounting for the presence of horizontal gene transfer (Martino et al., 2014). The potential influence and/or association of different environmental conditions with bacterial genetic features (Structure groups) was investigated through a stepwise forward approach using a multinomial logistic regression (MLR) analysis (Martino et al., 2011) and by the Fast UniFrac approach based on the concatenated sequences (Hamady et al., 2010).
Epidemiological analysis
Strain relationships were analyzed using PHYLOViZ (
Results
Sampling, isolation, and identification of V. parahaemolyticus strains
Isolates of Vibrio spp. (n = 160) were collected from the mollusks. Biochemical methods led to the classification of 140 putative V. parahaemolyticus strains, 102 of which were confirmed by species-specific gene amplification and MLSA. The summarized data on positive samples (31%) and on the identified strains are reported in Table 1, Figure 1, and Supplementary Table S1, respectively.
Molecular typing (MLST), virulence genes, and population structure
By applying the MLST scheme, 102 strains resulted in 63 unique STs, 49 of which (77%) were new in comparison with PubMLST database entries (Supplementary Table S2). It was not possible to attribute a ST for 24 strains due to incomplete amplification of the MLST (mostly recA and dtdS genes with 15/102 and 9/102 cases of nonamplification, respectively). The most frequent ST, ST481, represented 9% of analyzed strains. All genetic and environmental information regarding the strains isolated in this study has been deposited on the PubMLST web page (ID 1871–1972).
Regarding virulence analysis, no strains were found to be positive for the tdh gene, while six strains showed the presence of the trh gene.
Analysis of population structure and environmental and sampling data
The phylogenetic tree obtained by alignment of the concatenated sequences is shown in Figure 2a. Structure analysis revealed that strains formed two distinct populations (K = 2, ΔK = 330.66) as reported in Figure 2b. The first genetic population included 40 strains (S-A), whereas the second population comprised 24 strains (S-B). Strains having less than 90% of the characteristics of a specific group were included in a third mixed population (S-M, n = 38).
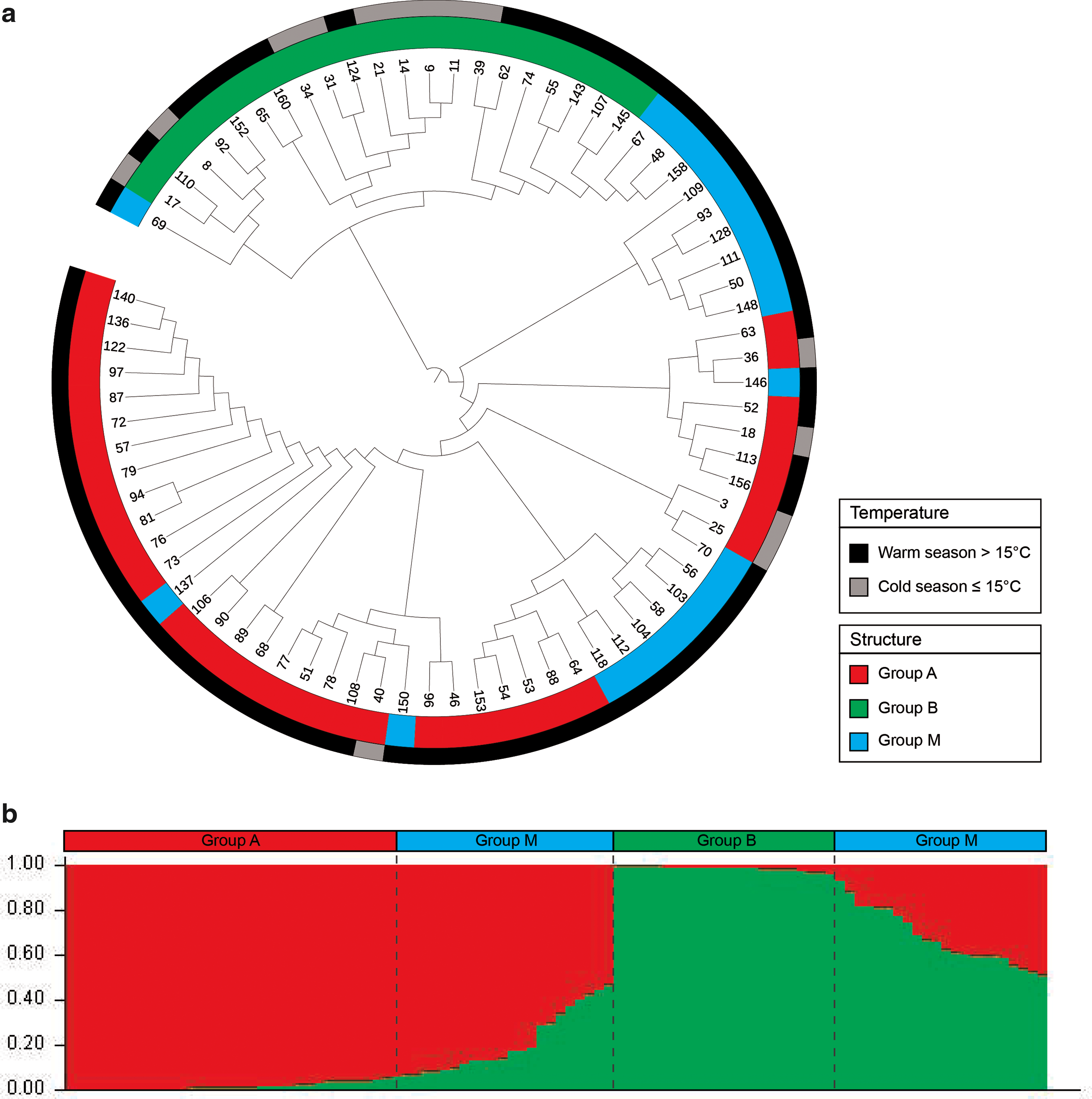
Phylogenetic and structure analysis
MLR analysis highlighted the influence of different environmental conditions on the distribution of the genetic populations defined by Structure. The stepwise procedure was used to select a model in which the origin of mollusks (production system: caught, reared, and unknown) and the temperature of sampling (cold ≤15°C/warm >16°C) were applied as predictors (p = 0.0001; R-Square Nagelkerke 0.304) of genetic structure. The contribution of each predictor was significant (p < 0.05); furthermore, the inclusion of the mollusk species (p > 0.05) in the model increased the classification performance (correct classification 59%). Observed and predicted frequencies of this model are reported in Figure 3.
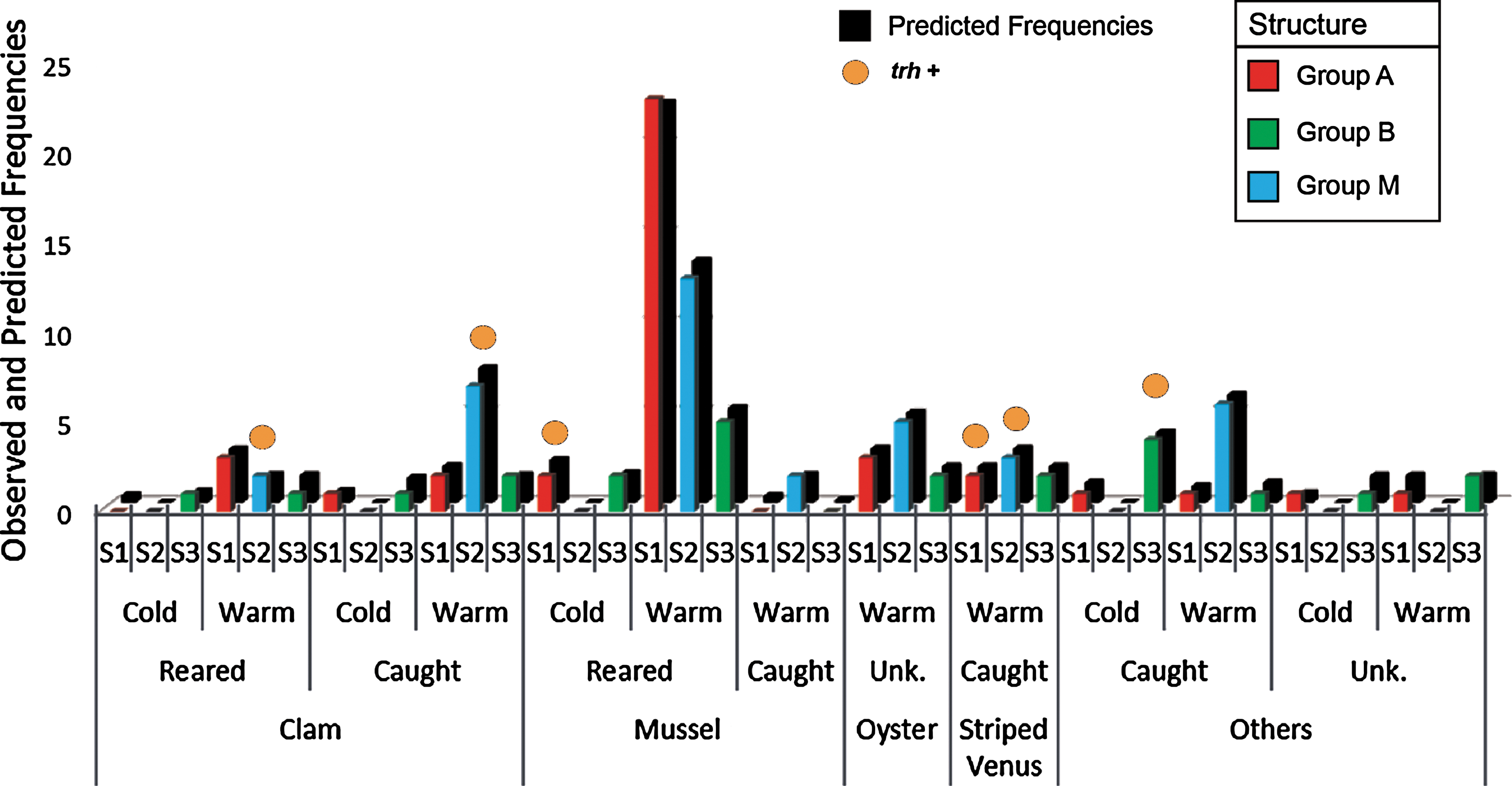
Structure group (S-A red; S-M blue; S-B green; n = 102 isolates) as per the full multinomial logistic model (warm/cold temperature, mollusk species, and production system). Predicted frequencies are reported in the dark bar. When origin appears as “unknown,” the system of production was not declared; these samples (Cerastoderma spp., P. jacobaeus, Crassostrea gigas etc.) were harvested from natural banks and from relaying areas. Color images available online at
The S-A genetic cluster was mainly associated with mussels reared during the warm period, while the strains ascribed to the mixed group (S-M) were the most representative of clams, striped venus (Chamelea gallina), and other species. A partial segregation was also observed according to the water temperature (cold/warm). The mixed group (S-M) was isolated only when the water temperature exceeded 15°C (Fig. 3). The S-B group was generally isolated during the cold period (<15°C), while 77% of the S-A strains were isolated at a temperature range over 21°C, as summarized in Supplementary Figure S3.
Regarding the virulence properties of the 102 V. parahaemolyticus, no association was found between trh+ strains and environmental parameters. However, the mollusk species that mainly hosted trh+ strains were clam and striped venus (Fig. 3). Two trh + isolates (31 and 124) were ascribed to ST470; these strains were collected from Pecten jacobaeus and C. gallina, respectively (Supplementary Table S2).
The unweighted UniFrac tests did not show significant differences in any of the comparisons performed. The presence of virulence genes as predictors highlighted a suggestive clustering of trh + strains (n: 40, 31, 150, 121) in comparison to trh − strains, as reported by the supported node in the jackknife tree (Supplementary Fig. S4 gray node). Conversely, the jackknife environmental clustering did not show specific patterns according to the temperature (Supplementary Fig. S4).
Analysis of CCs and epidemiological data
Strain relationships were analyzed using the PHYLOViZ program to identify potential CCs and founders. This analysis was compared to the PubMLST database to highlight potential relationships with strains isolated in other countries. Both the genetic data and the environmental and epidemiological information for all strains were considered. The PubMLST database presented 1520 MLST STs (STs) and 2254 available isolates at the date of analysis. The results, shown in Figure 4, suggest a partial segregation of S-A in relation to ST162 and ST481 as founders; however, the STs from mollusks were dispersed along all of the full MST branches. Most STs were singletons and did not display any relationship with the other isolates.
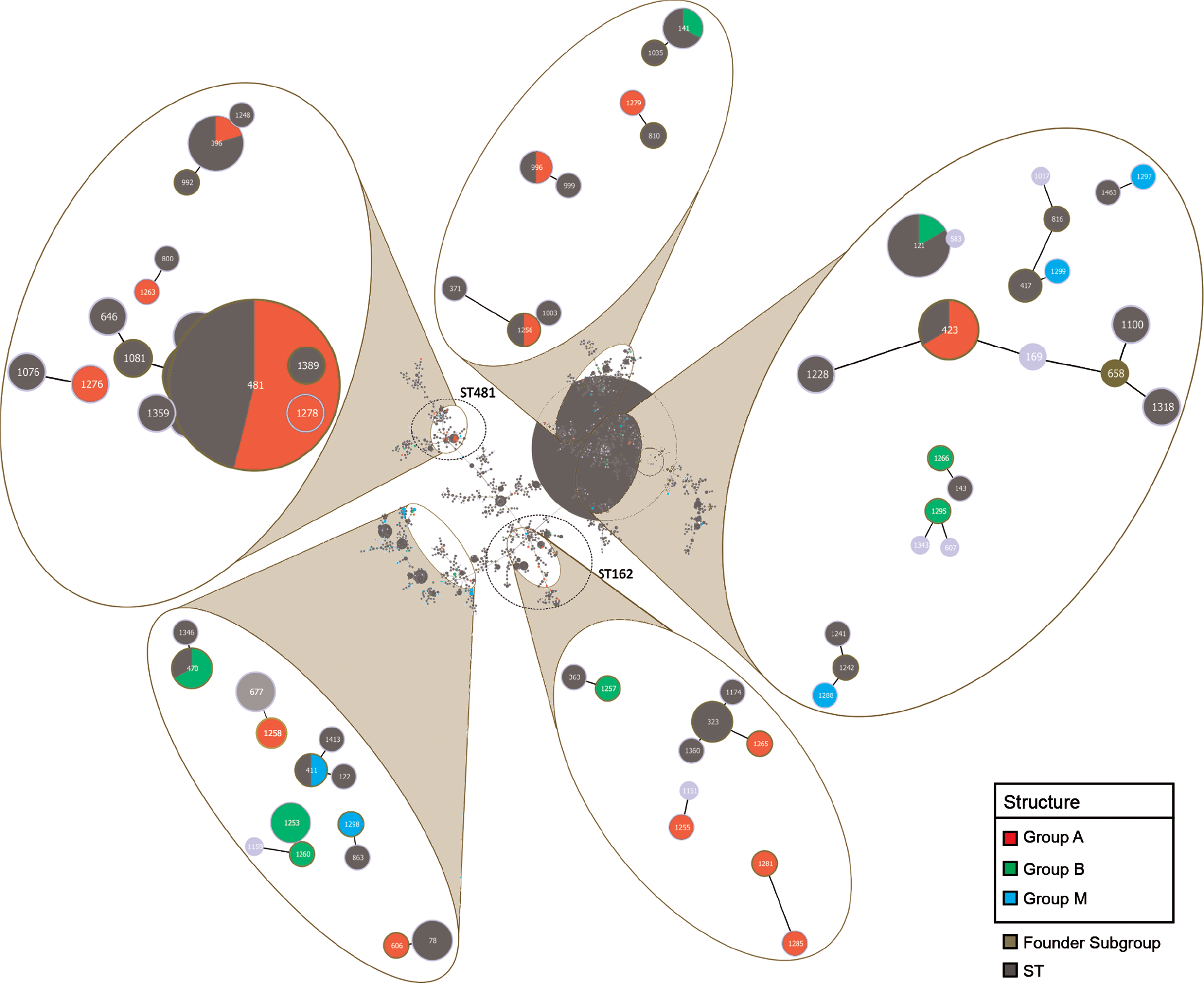
Full MST of all PubMLST database isolates; the sequence types (STs) collected form Italian samples were colored as per the structure populations (S-A red, S-M blue, and S-B green); the proportion of each node is related to the frequencies of each ST. The dotted circles highlight the ST162 and ST481. MLST, multilocus sequence typing. Color images available online at
Using the goeBURST algorithm, the strains collected during the survey on mollusks belong only to 25 clonal groups of the entire PubMLST dataset (Fig. 5). Table 2 summarizes the data analysis of the 25 CCs with the information provided by the PubMLST database. Figure 5 shows the clonal relationship between the Italian isolates and other worldwide STs. Interestingly, several STs are the same, or are closely related to STs isolated from Europe. Moreover, Figure 5 highlights some relationships between the strains isolated during the present survey and STs derived from shellfish collected at the polish market (Lopatek et al., 2015). The CC5 (Table 2) included these STs (ST481, ST1359) isolated from clams and reported to have Italian origins. Other STs are mainly related to strains isolated from Central Asia (Japan, China, and Korea). The inset of Figure 5 displays a selection of seven CCs that harbor virulence factors; the members of these CCs mainly displayed the trh gene and only the ST800 and ST816 presented the tdh gene.
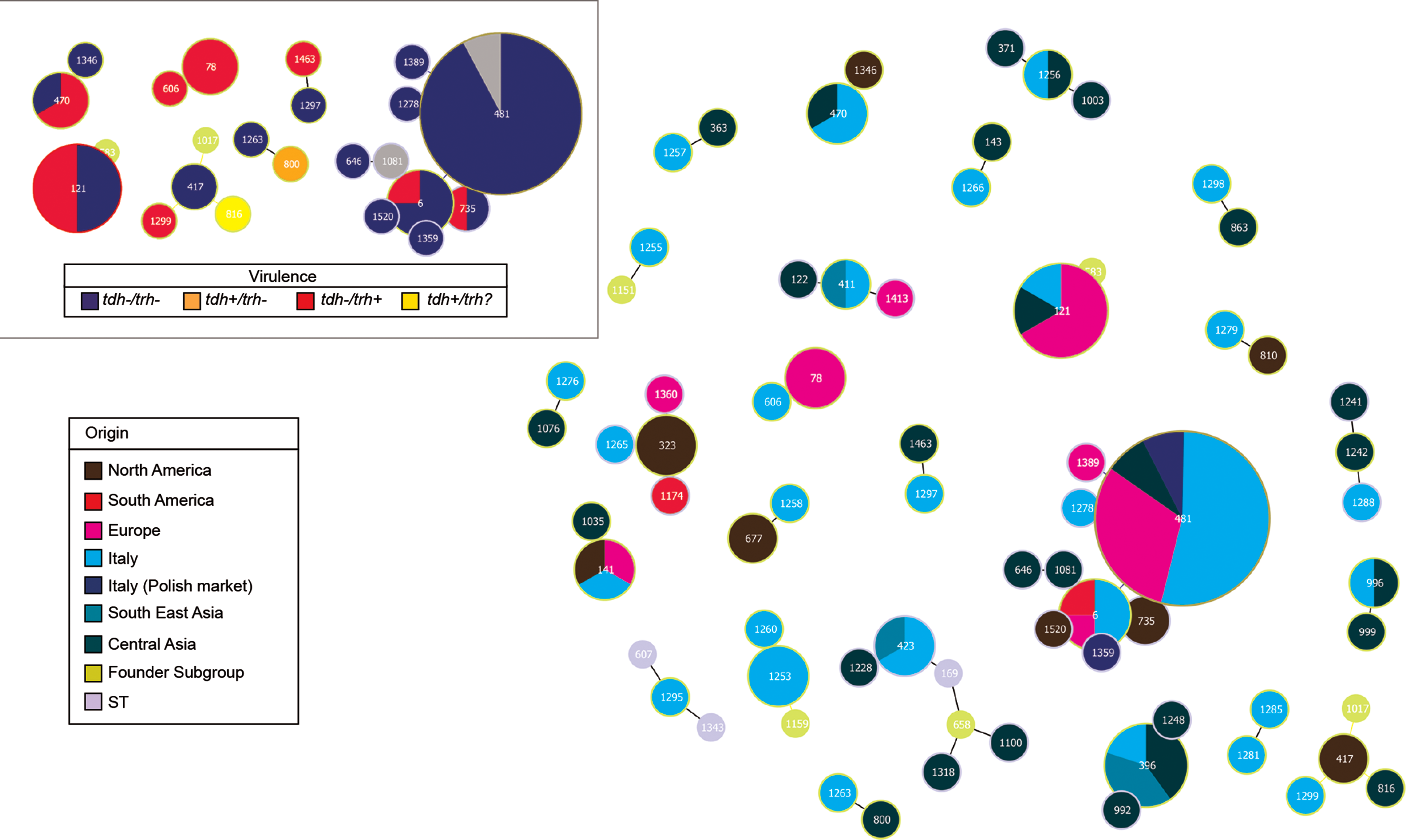
Clonal relationships among the STs collected from Italian samples and worldwide isolates from PubMLST, the inset elucidates the CCs of STs that harbored virulence factors. CCs, clonal complexes. Color images available online at
Italian STs are highlighted in bold.
CC = clonal complex/groups; numbers are referred to the default attribution of PHYLOViZ program.
Founder: the founder genotypes of the clonal complex/group.
Clinical generally resumed: gastroenteritis and watery diarrhea.
Seven CCs harbor virulence genes (PubMLST database), while also ST363 harbor tdh+/trh+ genes (Esteves et al., 2015). STs 800, 816, and 363 harbor the tdh gene.
MLST, multilocus sequence typing; ST, sequence type
Regarding the source of isolation, almost all the isolates had environmental or crustacean origin (Table 2). Six CCs showed genetic correlation between clinical STs involved in gastroenteritis and isolates from mollusks (Table 1). Merging the data on virulence factors and the STs isolated from gastroenteritis, only the ST1299 (CC33) showed a concomitant relationship between the clinical case and the presence of the trh virulence factor (Table 2). The presence of tdh/trh genes on the CC33 should be also carefully considered (Velazquez-Roman et al., 2014).
Discussion
Most studies of V. parahaemolyticus isolated from seafood are focused on the prevalence and distribution of these bacterial species, with a genetic characterization of the virulence genes (Suffredini et al., 2014; Caburlotto et al., 2016). Other studies aim at discovering the genetic relationships between environmental/food isolates and the strains involved in human illnesses (Yu et al., 2011; Gao et al., 2016).
MLST was demonstrated to be a reliable and simple tool to describe bacterial genetic diversity and to correlate it to the worldwide distribution of the V. parahaemolyticus populations (Urmersbach et al., 2014). The study of different genetic lineages in relation to detailed properties (e.g., habitat and geographic origin) is critical to obtain information on evolutionary relationships among isolates and to improve knowledge of the spread of different V. parahaemolyticus populations in certain areas (Urmersbach et al., 2014).
In this study, a detailed investigation was performed to characterize a representative number of V. parahaemolyticus strains (n = 102) from shellfish collected during a survey in the northeast of Italy.
Two clearly distinct genetic populations and a mixed group were discovered; these clusters were segregated per specific environmental features (water temperature), host (clams/mussels), and management systems (reared and caught). The S-B genetic group persisted at lower temperatures during the cold months, whereas the S-A and S-M groups were predominant during the warm season. These results highlighted not only that the composition of Vibrio species associated with mollusks varied according to water temperature (Rahman et al., 2014) but also that the genetic lineages of this hazardous species were probably subjected to a temperature selection. This agrees with Ellis et al., (2012), who discovered a strong correlation among genetic diversity and water temperature in the samples collected from the Great Bay Estuary.
The existence of genetic populations adapted to different water temperatures is extremely interesting with regard to global ocean warming and its impact on the spread and selection of waterborne pathogens (Vezzulli et al., 2016). A strong increase in genetic variability during the warm months was recently highlighted in strains collected from water in Mediterranean lagoons (Esteves et al., 2015). The 15°C threshold affected the Vibrio load (Baker-Austin et al., 2017) and the spread of pathogenic strains (Martinez-Urtaza et al., 2008). In this study, the presence of genetic populations adapted to water temperature (Cold ≤15°C >Warm) was also highlighted in shellfish products from the North Adriatic area, suggesting a strong adaptive global behavior of V. parahaemolyticus populations to temperature changes.
Overall, our data confirmed a worldwide spread of several STs; however, some CCs were present on the same continent (Europe) or country (Italy), suggesting a local dissemination of some STs. Interestingly, ST481 was also one of the most represented STs in the work of Esteves et al., (2015) and similarly isolated during warm months. Probably, this ST was associated with samples from Europe (Mediterranean and Baltic seas–Urmersbach et al., 2014) as also shown in the case of the Polish survey. Further comparisons with additional isolates could clarify if there is a maintenance of this ST core or a spread of new populations.
Few strains carried the trh + virulence factor (5.8%), whereas some clonal relationships enlarged the possibility of epidemiological links with clinical isolates (Esteves et al., 2015). The ST470 included other isolates from mollusks (Thailand-2003) and classified with serotype O1:KUT. As reported by Esteves et al. (2015), the ST363 was correlated with clinical isolates from Thailand. Other epidemiological links were also highlighted with clinical isolates from North American STs (ST417–CC33) (Han et al., 2014).
In comparison with the large number of isolates from mussels, clams hosted the most trh + strains. Several cases of clam-associated vibriosis were reported in the United States (Slayton et al., 2014). Further long-term studies are required to define the host role of mollusk species with respect to their different lifestyles. Moreover, the intrinsic properties of toxigenic strains could allow for the survival and spread of the pathogen (Velazquez-Roman et al., 2014).
The role of mollusks as a potential reservoir of different V. parahaemolyticus genetic populations and the high level of diversity observed (Esteves et al., 2015) could be considered as a background for the emergence of toxigenic strains. Further investigation of additional genes could shed light on the importance of isolates from mollusks as carriers of additional virulence profiles (Caburlotto et al., 2016).
Conclusions
The molecular characterization of V. parahaemolyticus provided a useful background on the genotype spread as a primary step in risk analysis and hazard identification. Moreover, these open data (first dataset of STs gathered from Italy) are available for further comparison as in the case of future outbreaks.
Local studies can elucidate particular behaviors of the Vibrio populations that could be useful to improve the management and safety of products by various stakeholders. The focus on specific hosts or different periods (cold ≤15°C >warm) could improve sample monitoring campaigns aimed at tools more tailored to the strains' variability.
In light of climate changes, the increase of warm-adapted genotypes should be considered during the formulation step of risk assessment. Moreover, the cold-adapted lineages could be a matter of further research on the resistance of V. parahaemolyticus.
Footnotes
Acknowledgment
The authors are also grateful to Ms. Morgan Smits (Department of Comparative Biomedicine and Food Science, University of Padova) for providing English language proofreading of the article.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.