
Abstract
Although infrequently associated with reported salmonellosis in humans, Salmonella enterica, subsp. enterica serovar Kentucky (ser. Kentucky) is the most common nonclinical, nonhuman serovar reported in the United States. The goal of this study was to use Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)–multi-virulence-locus sequence typing (MVLST) to subtype a collection of human clinical isolates of ser. Kentucky submitted to the Pennsylvania Department of Health and to determine the extent of antibiotic resistance in these strains. This analysis highlighted the polyphyletic nature of ser. Kentucky, and separated our isolates into two groups, Group I and Group II, which were equally represented in our collection. Furthermore, antimicrobial susceptibility testing on all isolates using a National Antimicrobial Resistance Monitoring System (NARMS) panel of antibiotics demonstrated that resistance profiles could be divided into two groups. Group I isolates were resistant to cephems and penicillins, whereas Group II isolates were resistant to quinolones, gentamicin, and sulfisoxazole. Collectively, 50% of isolates were resistant to three or more classes of antibiotics and 30% were resistant to five or more classes. The correlation of antibiotic resistance with the two different lineages may reflect adaptation within two distinct reservoirs of ser. Kentucky, with differential exposure to antimicrobials.
Introduction
N
The National Antimicrobial Resistance Monitoring System (NARMS) was established in 1996 among state health officials, the Centers for Disease Control and Prevention, the United States Food and Drug Administration, and the United States Department of Agriculture. One component of NARMS is performing antimicrobial susceptibility testing in Salmonella from human clinical isolates, retail meats, and food animals. In 2014, 57% of ser. Kentucky isolates from chickens in the United States were resistant to two to three classes of antimicrobials and 11% were resistant to four to five classes (NARMS, 2014).
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-based typing has been used in many different types of pathogenic bacteria, including Salmonella, Escherichia coli, and Campylobacter jejuni (reviewed in Shariat and Dudley, 2014). There are two CRISPR arrays in Salmonella, CRISPR1 and CRISPR2, and each comprises invariant direct repeat sequences of 29 nucleotides, separated by highly variable spacer sequences of 32 nucleotides (Touchon and Rocha, 2010). CRISPR spacer content is typically serovar specific as unrelated serovars rarely contain the same spacers (Fabre et al., 2012). Within a serovar, spacer duplication or deletion, or the presence of single-nucleotide polymorphisms (SNPs) allow for precise subtyping of isolates and has been demonstrated in multiple Salmonella serovars to date (Fabre et al., 2012; Shariat et al., 2015).
CRISPR-based approaches have been successfully used to subtype Salmonella, including outbreak isolates (Liu et al., 2011; Fabre et al., 2012; Shariat et al., 2013b; Li et al., 2014). CRISPR–multi-virulence-locus sequence typing (MVLST) combines a multilocus sequence typing (MLST) approach using sequence analysis of two virulence (MVLST) genes, fimH and sseL. Comprehensive analysis of four major serovars shown to cause illness (serovars Enteritidis, Heidelberg, Newport, and Typhimurium) demonstrated that CRISPR–MVLST and pulsed-field gel electrophoresis (PFGE) are comparable methods for strain discrimination (Liu et al., 2011; Shariat et al., 2013a, 2013b, 2013c).
Like some Salmonella serovars, such as ser. Newport, ser. Kentucky is polyphyletic (Sukhnanand et al., 2005; Sangal et al., 2010; Achtman et al., 2012; Timme et al., 2013; Haley et al., 2016), although the CRISPR patterns and antibiotic resistance profiles across both groups from isolates in the United States have not been investigated.
Our goal, in this study, was to use CRISPR–MVLST to subtype human clinical isolates of ser. Kentucky collected from Pennsylvania. We wanted to use this approach to examine strain diversity and determine whether antibiotic resistance correlates with subtyping analysis. Our results show that CRISPR-typing identifies two distinct groups, Groups I and II, reflecting ser. Kentucky polyphyly, and which exhibit differential patterns of antibiotic resistance.
Materials and Methods
Bacterial isolates
Forty clinical isolates of ser. Kentucky were obtained from the Pennsylvania Department of Health (Table 1), and comprise all ser. Kentucky isolates submitted to the Pennsylvania Department of Health between March 2007 and July 2015. The majority of isolates (24/40) was isolated from patients in southeastern Pennsylvania. After streaking on LB plates, single colonies were propagated overnight in 2 mL LB at 37°C. One milliliter of the culture was cryopreserved in 20% glycerol. Six hundred microliters of the culture was used to isolate genomic DNA using the Promega (Madison, WI) Genomic DNA Isolation Kit, following the manufacturer's directions. DNA pellets were resuspended in 200 μL of sterile water and stored at −20°C.
CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats; KST, Kentucky sequence type; MVLST, multi-virulence-locus sequence typing; PFGE, pulsed-field gel electrophoresis.
Polymerase chain reaction amplification and DNA sequencing
Polymerase chain reaction (PCR) amplification was performed on four genomic loci: CRISPR1, CRISPR2, fimH, and sseL, as previously described (Shariat et al., 2013b). Five microliters of each PCR was run on a 1% gel to confirm presence of a product. The remaining PCR was treated with exonuclease and Antarctic alkaline phosphatase (New England BioLabs) as previously described (Shariat et al., 2013b). Purified PCR products were sequenced at Eton Bioscience (Union, NJ). Some CRISPR1 and CRISPR2 arrays were longer (up to 2 kb) and necessitated additional sequencing primers designed to match internal spacers for sequencing. All primers used are indicated in Table 2.
(i), internal seq primer; PCR, polymerase chain reaction.
Sequence type analysis and pulsed-field gel electrophoresis
Sequences were assembled and aligned using SeqMan and MegAlign (Lasergene 13; Madison, WI). Both CRISPRs were further analyzed using CRISPR-finder (
PFGE using XbaI was performed using standard protocols (Ribot et al., 2006; Sandt et al., 2006).
Antibiotic resistance testing
Salmonella isolates were plated onto 5% sheep blood agar, and incubated overnight at 35°C. Suspensions equivalent to a 0.5 McFarland standard were prepared and adjusted to 1.0 × 105 colony-forming unit [CFU]/mL in Cation-Adjusted Mueller-Hinton Broth. The suspension was used to inoculate the CMV3AGNF panel (SENSITITRE™; Trek Diagnostics, Cleveland, OH) and incubated at 35°C for 18 h. All isolates were tested using a NARMS protocol for 14 drugs, amoxicillin, ampicillin, augmentin (amoxicillin/clavulanic acid) azithromycin, cefoxitin, ceftriaxone, ciprofloxacin, chloramphenicol, gentamicin, nalidixic acid, streptomycin, sulfisoxazole, tetracycline, and trimethoprim/sulfamethoxazole. Collectively, these 14 antibiotics represent 9 distinct classes of antimicrobials. Minimum inhibitory concentrations for drugs were tested and deduced based on defined breakpoints (NARMS, 2014; CLSI, 2015a, 2015b). A two-tailed t-test was used to determine significance between antibiotic susceptibility of the two ser. Kentucky groups. For multidrug resistance, intermediate resistance was not considered.
Results
CRISPR–MVLST and ST distribution
We used CRISPR–MVLST to subtype 40 ser. Kentucky isolates, which include all ser. Kentucky samples submitted to the Pennsylvania Department of Health over a 9-year period. Among these, we identified 13 CRISPR1 alleles, 8 CRISPR2 alleles, 2 fimH alleles, and 2 sseL alleles. CRISPR alleles were characterized by deletion or duplication of individual spacers, whereas allelic differences in fimH and sseL were defined by at least one SNP.
By combining the allelic variation at each of the 4 loci, we defined 14 unique ser. KSTs (Fig. 1). The three most frequent KSTs we identified were KST9 (11/40 isolates; 27.5%), KST1 (7/40; 17.5%), and KST10 (5/40; 12.5%). Eight KSTs were represented by single isolates. The calculated discriminatory power using CRISPR–MVLST was 0.8769.
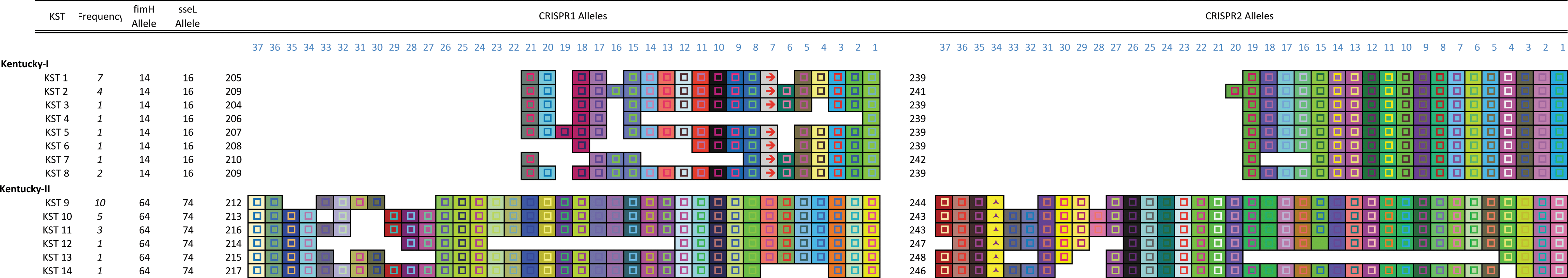
CRISPR–MVLST separates ser. Kentucky into two distinct groups. Graphic representation of the CRISPR arrays for CRISPR1 and CRISPR2 for the 14 ser. KSTs identified in this study. The number of isolates, out of the 40 analyzed, with a given KST is shown in italics. The number that identifies the fimH or sseL allele is shown and the number of the CRISPR allele is listed to the left of the respective CRISPR array. Each unique spacer is represented by a uniquely colored box, and the direct repeats are not shown. The spacers are aligned and the gaps represent the absence of a particular spacer. The direction of the spacers is from 5′ to 3′ with respect to transcription of the CRISPR loci. CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats; KST, Kentucky sequence type; MVLST, multi-virulence-locus sequence typing. Color images available online at
The CRISPR profiles (i.e., the spacer composition) of different Salmonella serovars are distinct. Within this collection of ser. Kentucky isolates, the CRISPR profiles separate the collection into two groups, Group I and Group II, with no single spacer found in both groups (Fig. 1). This analysis reflects the polyphyletic nature of this serovar, which is also observed in PFGE analysis of the same isolates (Fig. 2). The distinct composition of spacers within the CRISPR arrays demonstrates the separate evolutionary paths of the two groups. The two groups are evenly represented in our isolate collection: there are 18 Group I isolates (KST1-8) and 22 Group II isolates (KST9-14). There was no allelic overlap in any of the four loci across the two groups.

Polyphyly is reflected in PFGE analysis. PFGE using XbaI of all 40 isolates is able to separate the 2 groups of ser. Kentucky as indicated. PFGE, pulsed-field gel electrophoresis.
Isolates within each group possess a single allele for fimH and also for sseL, showing that for ser. Kentucky, these two alleles do not contribute to the subtyping scheme. Using Basic Local Alignment Search Tool (BLAST), we determined that the nucleotide identity between the two alleles of fimH was 1017/1035 (98%) and 968/981 (99%) between the two sseL alleles.
BURST (Based Upon Related Sequence Types) analysis demonstrates similarities between MLST groups, so we applied this to our CRISPR–MVLST analysis. We showed that KST1–6, and −8 group fall into a single BURST group (BURST Group1), and that KST10 and −12 fall into BURST Group2 (Supplementary Fig. S1; Supplementary Data are available online at
Antimicrobial susceptibility testing
We sought to determine the antibiotic resistance profiles for all 40 isolates in our collection. Among the 14 antibiotics tested, we observed broad resistance against tetracycline (56% of isolates), ampicillin (48%), and streptomycin (43%) (Fig. 3a). A single isolate was resistant to chloramphenicol and trimethoprim–sulfamethoxazole (M10028384001A; KST9) and all isolates were susceptible to azithromycin. Interestingly, resistance profiles against additional antibiotics correlate with the polyphyletic group. Isolates from Group I are resistant to amoxicillin, cefoxitin, and ceftriaxone and show intermediate resistance to augmentin, whereas Group II isolates are sensitive to these antibiotics. Conversely, Group II isolates show resistance to nalidixic acid, sulfisoxazole, gentamicin, and ciprofloxacin, whereas Group I isolates are largely susceptible to these antibiotics (Fig. 3b). The antibiotic resistance profiles for individual isolates are shown in Supplementary Table S1. In total, 50% of the ser. Kentucky isolates (20/40) submitted to the Pennsylvania Department of Health during 2007–2015 exhibited resistance to more than three classes of antibiotics. Of these, 60% were resistant to five or more classes of antibiotics (Supplementary Table S1). Importantly, 10/11 KST9 isolates (representing 25% of all isolates) are resistant to ciprofloxacin. Only a quarter of the strains (10/40) were susceptible to all antibiotics tested.
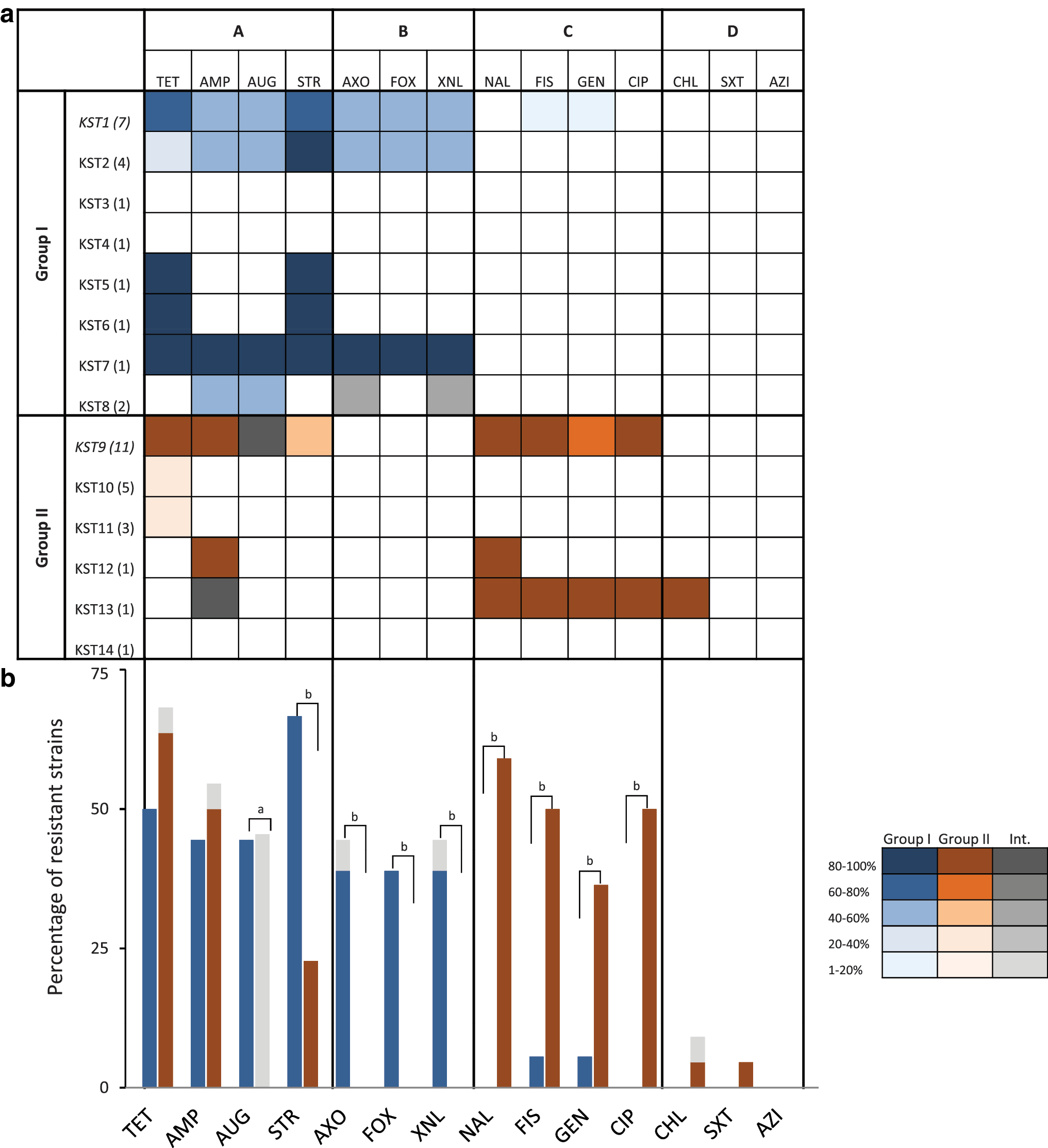
Antibiotic resistance correlates with phyletic group.
Correlation with MLST studies
A seven-gene MLST scheme is routinely used to type Salmonella and two common ser. Kentucky STs include ST152 and ST198 (EnteroBase; Available at:
ST198 and ST152 do not have any allelic overlap (0/7 alleles), similar to the CRISPR–MVLST data, where 0/4 alleles overlap across the two Groups I and II.
Discussion
Data presented in this study shows that CRISPR–MVLST is able to separate 40 ser. Kentucky isolates into 14 distinct KSTs, KST1–KST14. The majority of strain diversity is resolved by analysis of the CRISPR1 locus, which shows the greatest polymorphisms across different strains. As seen in other Salmonella serovars, neither CRISPR locus appears to be adapting through acquisition of new spacers (Shariat et al., 2015). Rather, allelic differences arise from deletion or duplication of individual spacer-repeat units. These data are comparable to CRISPR–MVLST analysis of other serovars (also human derived), for example, analysis of 141 ser. Enteritidis isolates generated 22 Enteritidis STs, 89 ser. Heidelberg isolates generated 21 Heidelberg STs, and 86 ser. Typhimurium generated 37 Typhimurium STs (Shariat et al., 2013a, 2013c).
Serovar Kentucky is polyphyletic (Sukhnanand et al., 2005; Achtman et al., 2012; Timme et al., 2013; Haley et al., 2016) and both phyletic groups are equally represented in our collection of isolates, which consists of all ser. Kentucky isolates that were submitted to the Pennsylvania Department of Health over a 9-year period. We termed these two lineages Group I and Group II. KST1 (17.5% of all isolates) and KST9 (27.5%) were the two most frequent types of each group. Whole genome sequence data of multiple ser. Kentucky isolates have shown that the two groups bear less resemblance to each other than to other Salmonella serovars (Timme et al., 2013; Haley et al., 2016), and this is reflected in our allelic data, where no alleles are shared between the two groups.
Antibiotic susceptibility testing highlighted differences between Groups I and II. While many isolates exhibit resistance to the antibiotics tetracycline (58% of isolates), ampicillin (50%), streptomycin (43%), and intermediate or full resistance to augmentin (45%), there were surprising differences when the groups were considered separately. Group I isolates exhibited resistance against amoxicillin and two cephems (cefoxitin and ceftriaxone) and are largely resistant to cephems and penicillins. Conversely, Group II isolates are resistant to quinolones (nalidixic acid and the fluoroquinolone, ciprofloxacin), gentamicin, and sulfisoxazole. The fact that half the isolates in our collection showed multidrug resistance to three or more classes of antibiotics complicates treatment options in patients with invasive ser. Kentucky infections.
Other studies examining ser. Kentucky have used MLST analysis to identify two common STs, ST198 and ST152. Our in silico data, using genome sequences available on National Center for Biotechnology Information (NCBI) (Haley et al., 2016) (Supplementary Fig. S2), show that all ST152 isolates (using 13 consensus sequences that were derived from 101 individual genomes) were in CRISPR Group I and that ST198 isolates (from 2 consensus sequences/9 genomes) fall into Group II.
ST152 isolates are responsible for the majority of ser. KSTs identified in nonhuman samples in the United States (Le Hello et al., 2011; Haley et al., 2016). In a study using whole genome sequencing of 112 poultry and cattle-derived isolates in the United States, 90% were ST152/Group I (Haley et al., 2016). Furthermore, all (31) isolates analyzed in that study from Pennsylvania were Group I (29/31 were ST152, 2/31 were ST318), so it seems that in Pennsylvania at least, the ST152/Group I reservoir is in domestic food animals.
Conversely, in that study, only 5% of nonhuman isolates were typed as ST198/Group II, showing that this ST is infrequently associated with poultry and cattle in the United States. Enterobase also shows low levels of ST198 associated with food animals (EnteroBase). While our sample size, 40, is small (we typed all ser. Kentucky isolates in the Pennsylvania Department of Health collection), the frequency of Group I to Group II infections is comparable (45–55%, respectively), suggesting that there may be an alternative reservoir for Group II/ST198 that does not exist in domestically produced food.
Multiple studies have linked ST198 infections with international travel to North Africa and the Middle East, where ST198 is prevalent in poultry (Le Hello et al., 2011, 2013; Mulvey et al., 2013). More recently, ser. Kentucky cases have been linked to travel to India, as well as domestically within Europe (Le Hello et al., 2013). In the United States, five ST198 infections were related to imported spices from North Africa between 2002 and 2009 (Le Hello et al., 2013).
A comprehensive study by Le Hello et al. (2011) subtyping human clinical ser. Kentucky isolates from France, England and Wales, Denmark, and the United States show that 74% of isolates were ST198 and that over a quarter of these were resistant to ciprofloxacin. This study implicates international travel or import of contaminated food as causes for ST198 infection in North America, and while ST198/Group II isolates are present at low levels in food animals in the United States, it is not clear whether there is another, larger, domestic reservoir for Group II (ST198).
Unlike serovars Enteritidis and Heidelberg, which are frequently isolated from both poultry and humans, ser. Kentucky is the leading serovar identified in chickens in the United States, and is also found in cattle, yet it is rarely found to cause illness in humans. This suggests some limitation to its pathogenicity in humans. Perhaps the ST152/Group I isolates do not cause severe symptoms in healthy individuals, thus, fewer cases are reported. That ST198/Group II Salmonella are found at lower levels in domestic food animals may be because its colonization is limited by the presence of ST152/Group I isolates. It is possible that although ST198 is infrequently found in nonhuman samples in the United States, that when human infections do occur, they are more severe than ST152, and thus greater number of patients report their illness. Serovar Kentucky ST198 is more commonly found to infect humans elsewhere in the world, suggesting that this strain is indeed more infectious, although this strain is also found internationally in poultry. Continual monitoring of our food supply animals for ST198/Group II isolates, combined with antimicrobial susceptibility testing should be prioritized.
The phenotypic and genetic differences between Group I and II ser. Kentucky isolates presented in this study highlight their distinct lineages and suggest they inhabit two distinct ecological reservoirs. Future investigations may address the following outstanding questions: (1) whether Group I (ST152) isolates are better adapted to poultry and cattle than Group II (ST198) isolates (at least in the United States), (2) whether there are other domestic reservoirs of ser. Kentucky, especially since the incidence of Group II is much lower in poultry and cattle, (3) to determine the extent of selective pressure from antibiotics in the different reservoirs, and (4) to determine whether genetic differences in Group II ser. Kentucky are responsible for increased prevalence in causing illness.
Succinctly, our findings show that the evolutionarily distinct Groups I and II of ser. Kentucky have both caused illness in humans in Pennsylvania and that the antibiotic resistance profiles are different between the two lineages. These findings may have implications to other polyphyletic Salmonella, including serovars Newport, Muenchen, Seftenberg, Bareilly, and Paratyphi B (Timme et al., 2013).
Footnotes
Acknowledgments
The authors thank Kelly Kline and members of the Shariat Laboratory for critically reviewing this article, and Drs. Shelly Rankin and Zakiya Whatley for helpful discussions. Funding was provided by Gettysburg College (startup funds to N.S.), the United States Department of Agriculture-National Institute of Food and Agriculture (grant 2016-69003-24615 to N.S.), and the Commonwealth of Pennsylvania and the NARMS through a cooperative agreement (CDC ELC-04040). The authors also thank Dr. Phillipe Horvath (Danisco) for providing them with the Macro to visualize CRISPR spacers.
Disclosure Statement
The authors state that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.