
Abstract
Campylobacter has been the most commonly reported cause of bacterial diarrheal disease in humans in the European Union since 2005. Most broiler batches at slaughter are colonized with Campylobacter, and the major source of infection is contaminated poultry meat. The aim of this study was to characterize a selection of Campylobacter jejuni and Campylobacter coli isolates from broilers through whole-genome sequencing (WGS). A total of 16 isolates (C. jejuni = 12 and C. coli = 4) from five broiler farms from Catalonia (northeastern Spain) were analyzed. A phylogenetic analysis based on 8420 single-nucleotide polymorphisms showed two main cluster grouping strains by species. Phenotypic resistances to quinolones (100%), tetracycline (81%), streptomycin (75%), erythromycin (56%), and gentamicin (13%) were found. All the isolates carried the C257T point mutation in the subunit A of the DNA gyrase gene (Thr86Ile) conferring resistance to quinolones, while all the isolates showing resistance to tetracycline carried the tet(O) gene. The genes aph(3′)-III and aadE conferring resistance to aminoglycosides were identified in the two isolates (one C. jejuni and one C. coli) resistant to streptomycin and gentamicin. The point mutation A2075G on the 23S rDNA conferring high resistance to macrolides was detected in three C. coli isolates. The CmeABC multidrug efflux pump was also detected, both in C. jejuni and C. coli isolates. All C. jejuni and C. coli isolates were positive for most of the 34 virulence-associated genes studied related to motility, chemotaxis, adhesion, and invasion. Interestingly, the wlaN gene involved in the Guillain–Barré syndrome was found in two isolates. The results underline the power of WGS for investigation of virulence, clonality, and antimicrobial resistance in Campylobacter.
Introduction
S
In the majority of the EU countries, most of the broiler batches are colonized with Campylobacter at slaughter and the main source of campylobacteriosis in humans is chicken meat, which can account for up to 70% of cases (Boysen et al., 2014). The prevention of broiler flock colonization has therefore become a food safety priority in the European Union (EFSA, 2011), reflected by the new regulation (amendment of Annex I to EC regulation No. 2073/2005 with regard to Campylobacter in broiler carcasses) that may enter into force in 2018.
The pathogenicity of Campylobacter strains has been linked to multiple factors, including host susceptibility and, more importantly, the expression of different virulence factors and resistance to antimicrobials. Several putative virulence factors have been identified in Campylobacter species that contribute to motility, intestinal adhesion, colonization, toxin production, and tissue invasion (Dasti et al., 2010; Bolton, 2015). Also, multidrug-resistant C. jejuni and C. coli have been reported worldwide from farm animals and retail meats, including poultry and swine (Datta et al., 2003; Zhao et al., 2010).
Phenotypic methods have been widely used to characterize C. jejuni and C. coli strains. However, these methods have mostly been replaced by genotypic methods that are more accurate and have higher discrimination power, such as pulsed-field gel electrophoresis (PFGE) and multilocus sequence typing (MLST) (Pfaller, 1999). Nevertheless, with the advent of next-generation sequencing, the possibility of generating high-resolution full genome data is being increasingly used to differentially characterize strains. This technology allows for a rapid identification of a broad range of genotypic traits of the isolates, such as their pool of virulence and antimicrobial resistance determinants. It has proven useful in gaining insight into the epidemiology of Campylobacter and predicting its antimicrobial resistance (Zhao et al., 2015; Llarena et al., 2017).
Hence, the aim of this study was to take advantage of whole-genome sequencing (WGS) to in-depth characterize a subset of C. jejuni and C. coli isolates from broilers obtained from a longitudinal study involving different farms. The characterization included the determination of the MLST genotype, the identification of virulence and antimicrobial determinants, as well as a phylogenetic study of the isolates through the discovery of single-nucleotide polymorphisms (SNPs) between the different strains analyzed.
Materials and Methods
Isolates
A total of 16 poultry isolates (C. jejuni = 12 and C. coli = 4) from 5 broiler farms were included in the study. The isolates were selected from Campylobacter-positive flocks of a broad 2-year longitudinal study (2011–2013), where six to seven flocks were studied each year by cloacal swab sampling a subset of birds. Selection of the isolates was performed according to their PFGE patterns (Supplementary Fig. S1; Supplementary Data are available online at
Campylobacter isolation and identification were performed as previously described (Urdaneta et al., 2015). Isolates were preserved in a brain–heart infusion broth (Merck KGaA, Darmstadt, Germany), with 20% glycerol at −80°C until used, and fresh cultures of the isolates were prepared on Columbia blood agar plates (bioMérieux, Marcy-l'Etoile, France). Plates were incubated at 37°C for 48 h under microaerobic conditions using a microaerobic atmosphere generator (Anaerocult® C; Merck, Darmstadt, Germany).
Antimicrobial susceptibility testing
Isolates were tested for antimicrobial susceptibility using a minimum inhibitory concentration (MIC)-based broth microdilution (VetMIC GN-mo; National Veterinary Institute, Uppsala, Sweden) for the following antimicrobial agents: nalidixic acid (1–64 mg/L), ciprofloxacin (0.06–8 mg/L), tetracycline (0.12–16 mg/L), streptomycin (0.5–64 mg/L), gentamicin (0.12–16 mg/L), and erythromycin (0.5–64 mg/L). C. jejuni ATCC 33560 and C. coli ATCC 33559 were used as control strains. An isolate was considered multidrug resistant when showing resistance to three or more nonrelated antimicrobials. Isolates were considered to be susceptible or resistant based on epidemiological cutoff values according to EUCAST guidelines (
WGS and assembly
Genomic DNA was extracted using QIAamp DNA mini kit (QIAGEN) according to the manufacturer's instructions. The libraries were prepared with Nextera XT DNA sample preparation kit (cat. no. FC-131-1024; Illumina, Inc., San Diego, CA) followed by multiplexed paired-end sequencing with a read length of 2 × 251 bp, using Illumina's MiSeq platform (Illumina).
The raw reads were trimmed and cleaned for adapters, and assembling was performed using the online tool Assembler v1.2 with default parameters. All these steps are integrated in a pipeline available at the Center for Genomic Epidemiology (CGE) (
The raw sequence data set is available in the NCBI database with BioProject accession number PRJNA385807.
Analysis of resistance and virulence-associated genes
ResFinder v2.1 (
All strains were subjected to analysis of the presence of resistance determinants to quinolones, tetracyclines, aminoglycosides, and β-lactams. C. jejuni and C. coli strains were tested for 34 virulence-associated genes; the identifiers of each of the genes analyzed and the homology analyses are detailed in Supplementary Tables S1 and S2, respectively. The presence of several virulence-associated genes related to motility (eight), chemotaxis (five), adhesion (four), invasion (three), cytolethal distending toxin (three), multidrug and bile resistance (three), stress response and survival (two), iron uptake (two), capsule (two), Guillain–Barré syndrome (one), and hippuricase (one) was assessed (Koolman et al., 2015). All genes were identified with a selected identity threshold of 80% (Zankari et al., 2012) and a minimum coverage of 20% of the query sequence length. The presence of the flaA and flaB motility genes was confirmed by PCR with specific primers (Koolman et al., 2015).
To analyze the presence/absence of specific mutations related to antibiotic resistance, the raw FASTQ files for each of the isolates were aligned with the BWA-MEM algorithm (Li and Durbin, 2009) with the corresponding reference genome (AL111168 for C. jejuni and CP011015 for C. coli). The alignment files and the corresponding annotated reference genome were inspected manually using Tablet as the visualizing tool (Milne et al., 2013). Only mutations that appeared with frequency higher than 0.5% were considered.
Identification of SNPs
The SNP discovery was done using the CSI Phylogeny v1.4 pipeline CGE (Kaas et al., 2014). Briefly, the paired-end reads from each of the isolates were reference aligned using strain 12 for C. jejuni and 4 for C. coli with BWA v.0.7.2 software (Li and Durbin, 2009). SNP calling was done with SAMtools v.0.1.18 [“mpileup” method; Li and Durbin (2009)] and filtering was done with BEDTools (Quinlan and Hall, 2010). To select the valid SNPs, the same criteria previously described were followed (Kaas et al., 2014). SNPs were filtered out if the mapping quality was below 25 or the SNP quality was below 30, and if they were called within the vicinity of 10 bp of another SNP (pruning).
To perform the phylogenetic analysis, concatenation of the sequences and subsequent aligning with MUSCLE were done (Edgar, 2004). Maximum likelihood trees were created using CSI Phylogeny 1.4 (available at
Multilocus sequence typing
The de novo assembled contigs were used to identify the multilocus sequence types (STs) and clonal complexes (CC) using the web server MLST v1.8 available at the CGE website (
Results
Whole-genome sequencing
The isolates were sequenced to an average coverage that varied from 49.73 to 127.2 × (Supplementary Table S3). From the assembled contigs of the 16 isolates, 9 previously described STs were recognized (Fig. 1). Besides, two novel STs not previously reported in the PubMLST database (
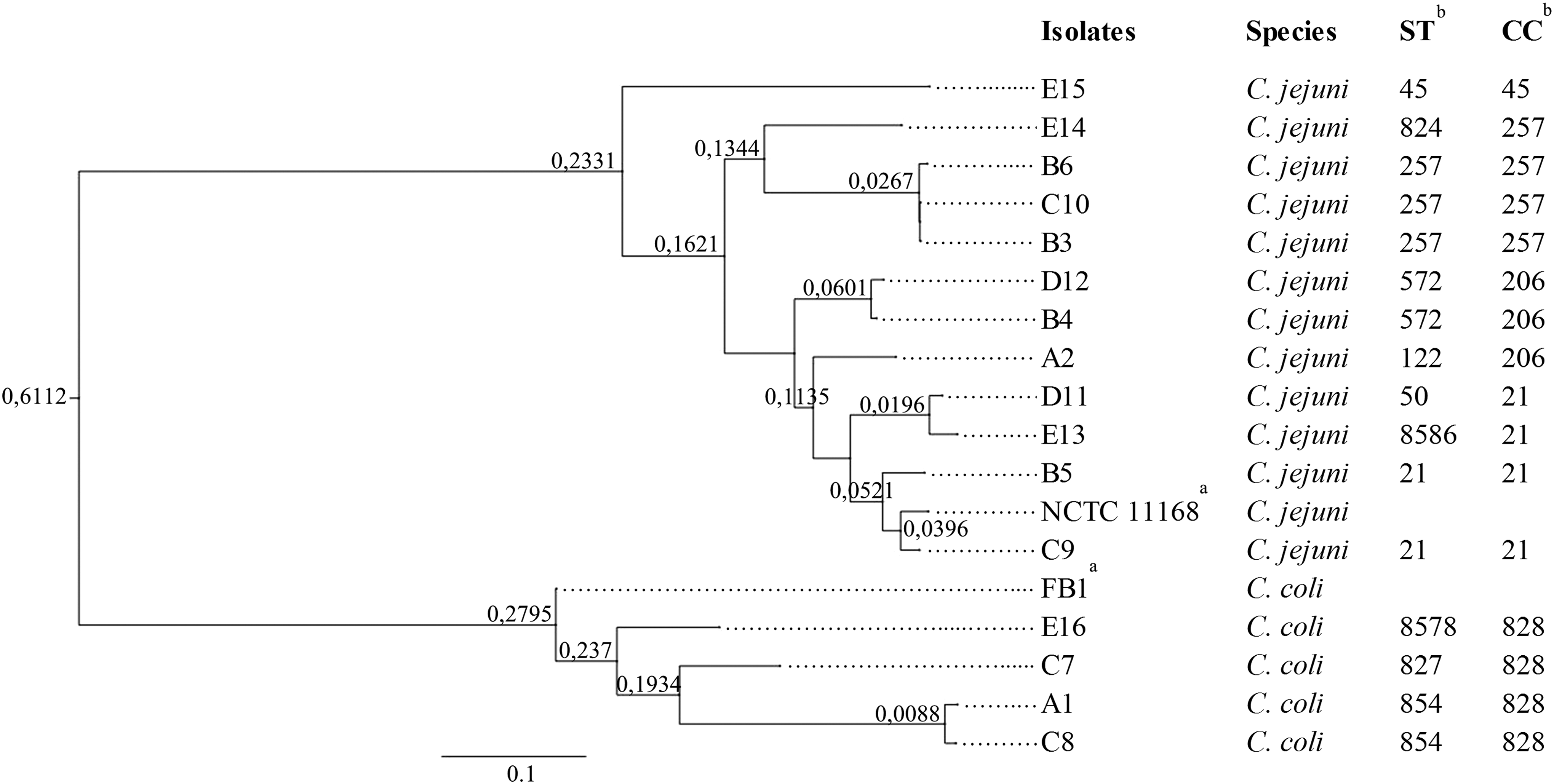
Maximum-likelihood phylogenetic tree based on SNPs from the assembled genomes and genotypic MLST types of the Campylobacter isolates. The tree was drawn to scale, with branch lengths measured in the number of substitutions per site. aReference genomes for Campylobacter jejuni NCTC 11168 (accession no.: AL111168) and for Campylobacter coli FB1 (accession no.: CP011015) were included in the analysis. bST, sequence type; CC, clonal complex; SNP, single-nucleotide polymorphism.
Comparison of the sequences obtained from the 16 isolates allowed the identification of a total of 8420 SNPs, which were used to perform the phylogenetic analysis. The representation of the SNP tree is depicted in Figure 1 and a heatmap representing the SNP counts between the genomes is shown in Supplementary Figure 2. None of the isolates analyzed was identical. The isolates showed distribution in two main clusters, with all strains grouped by species.
Antimicrobial susceptibility
The phenotypic antimicrobial susceptibility patterns determined for the 16 strains analyzed are shown in Table 1. All the strains showed a multidrug-resistant profile, with resistance to quinolones (nalidixic acid and ciprofloxacin) being common to all of them. The resistance to tetracycline was the second most commonly observed (81%) followed by streptomycin and erythromycin resistance (75% and 56%, respectively). Among the latter, one-third of C. jejuni isolates were resistant, while all C. coli isolates showed resistance to this antimicrobial. Gentamicin resistance was the less prevalent, detected only in two isolates from farms A and B (13%).
R-mech: resistance mechanism. Thr86Ile: point mutation in the subunit A of the DNA gyrase gene; tet(O), aphA(3′), and aadE: presence of these genes; 23S rDNA: point mutation on this region of the genome.
Interpretation of MIC values for C. jejuni epidemiological cutoff values: Nal (R ≥ 16 mg/L); Ci (R ≥ 0.5 mg/L); Tc (R ≥ 1 mg/L); Sm (R ≥ 4 mg/L); Gm (R ≥ 2 mg/L); and Ery (R ≥ 4 mg/L). Interpretation for C. coli epidemiological cutoff values: Nal (R ≥ 16 mg/L); Ci (R ≥ 0.5 mg/L); Tc (R ≥ 2 mg/L); Sm (R ≥ 4 mg/L); Gm (R ≥ 2 mg/L); and Ery (R ≥ 8 mg/L).
Ci, ciprofloxacin; Ery, erythromycin; Gm, gentamicin; MIC, minimum inhibitory concentration; Nal, nalidixic acid; Sm, streptomycin; Tc, tetracycline.
To study the potential mechanisms of antimicrobial resistance, the assembled genomes of each of the strains under study were investigated for particular patterns known to be associated to resistance. Phenotypic antimicrobial resistance obtained with the MIC analysis corresponded well for most of the isolates with the identification of specific antimicrobial resistance genes detected by WGS (Table 1). All the isolates carried the C257T point mutation in the subunit A of the DNA gyrase gene (Thr86Ile) conferring resistance to quinolones. Other less common mutations in gyrA (Asp-90-Asn and Ala-70-Thr) were not detected in any of the isolates. All the isolates showing resistance to tetracycline carried the tet(O) gene. Within the two isolates resistant to streptomycin and gentamicin, the genes aph(3′)-III and aadE conferring resistance to aminoglycosides were identified. Three C. coli isolates were found to show a mutation in the A2075G position of the 23S rDNA region, which confers a high level of resistance to macrolides. The CmeABC multidrug efflux pump has been described as the major efflux pump mechanism conferring resistance to a wide range of antimicrobials, and it was identified in 15 out of the 16 isolates analyzed.
Virulence determinants
All isolates of C. jejuni and C. coli were positive for almost all of the 34 virulence-associated genes studied, including motility, chemotaxis, adhesion, and invasion genes, with few exceptions (Table 2). The flagellin genes were unexpectedly found absent in most of the strains through WGS analysis. However, due to the known difficulty associated to accurately assemble duplicated genes, the presence of these flaA and flaB genes was assessed by PCR. Table 2 shows the experimentally validated presence of flaB in all the isolates and absence of flaA in one-third of the isolates by specific PCR. Only one isolate was negative for cmeB (component of the CmeABC efflux pump) and another one for cfrA (gene involved in iron uptake), both were C. jejuni strains from farm E. Remarkably, the wlaN gene, involved in the Guillain–Barré syndrome, was detected in two C. jejuni isolates from different farms. As expected, the hipO gene was not detected in any C. coli isolate.
The presence of genes related to motility (flhA, flhB, flgB, flgE, fliM, fliY); chemotaxis (cheA, cheB, cheR, cheW, cheY); adhesion (cadF, dnaJ, pdlA, racR); capsule (kpsM, waaF); invasion (iamA, ciaB, ceuE); cytolethal distending toxin (cdtA, cdtB, cdtC); stress response and survival (katA, sodB) was also confirmed in all the isolates (data not shown to facilitate the reading).
The results for fla genes were confirmed by PCR.
Discussion
The whole-genome sequence data revealed that the C. jejuni and C. coli isolates belonged to five different CC, all of them associated both with poultry and human campylobacteriosis in many countries (Campylobacter PubMLST;
The comparison of the assembled genomes revealed a large number of nucleotide changes (SNPs), among the isolates and the reference genomes. The identified variations were used to study the phylogeny to infer the relationship among the isolates, which were in concordance to the species they belong (Fig. 1). Not surprisingly, isolates of identical STs were more closely related compared with isolates of different STs.
The antimicrobial resistance found is of relevance in a public health context, particularly those to fluoroquinolones and macrolides (mainly ciprofloxacin and erythromycin, respectively), and also tetracyclines and aminoglycosides. Quinolones and macrolides are the antibiotics of choice to treat severe human Campylobacter infections, while tetracyclines are used as an alternative treatment (Butzler, 2004; Moore et al., 2006) and aminoglycosides are recommended to treat bacteremia caused by Campylobacter (Kassa et al., 2007).
Resistance to fluoroquinolones and macrolides is quite common in poultry (Thakur et al., 2010). High quinolone resistance in poultry has previously been reported in Spain (Melero et al., 2012; Pérez-Boto et al., 2013), as well as in other EU countries (Luber et al., 2003; Nobile et al., 2013). The high MIC values detected here may be related to the presence of Thr86Ile in all of the isolates, which in itself provides high resistance to quinolones (MIC >16 mg/L) (Ruiz et al., 1998). This mutation is the most prevalent in clinical and veterinary isolates (Butzler, 2004; Hormeño et al., 2016). The high prevalence of isolates resistant to tetracyclines is similar to that previously reported in poultry in Spain (Melero et al., 2012; Pérez-Boto et al., 2013; Duarte et al., 2016). This resistance is mediated by the tet(O) gene, which was detected in all isolates showing tetracycline resistance. Besides the chromosomal location of this gene, it has also been reported in plasmids (Avrain et al., 2004; Iovine, 2013). In contrast, resistance to aminoglycosides was diverse, with a considerably high resistance to streptomycin (75%) and a much lower resistance to gentamicin (13%), in agreement with Duarte et al. (2016) and Pérez-Boto et al. (2013). Gentamicin resistance in Campylobacter spp. from poultry is a rare event all over European countries (De Jong et al., 2009; Carreira et al., 2012; Pérez-Boto et al., 2013), probably because it is not used in poultry production. The aph(3′)-III and aadE genes involved in aminoglycoside resistance were identified in some strains. However, the finding of high resistance to streptomycin in some of the strains despite the absence of corresponding genes might be due to the presence of undiscovered genes. Nevertheless, the present work was focused on chromosomal genes, and so, whether these strains carried plasmids encoding streptomycin resistance genes cannot be ruled out and deserve further research (Iovine, 2013). Over 50% of isolates were resistant to erythromycin and more common among C. coli than C. jejuni, similarly to what has been previously reported in poultry in the European Union (Wimalarathna et al., 2013; Duarte et al., 2016). Erythromycin resistance is acquired through point mutations in domain V of the 23S rDNA at positions 2074 and 2075 (positions 2058 and 2059 in E. coli numbering) (Iovine, 2013); the point mutation A2075G, which is the most prevalent in Campylobacter spp. and confers high-level resistance to macrolides, was identified in three C. coli isolates. The overall resistances detected here are those also common in food-producing animals in the European Union, as reported by EFSA (2015). Particularly, the pattern of resistance among Campylobacter isolates was predominantly quinolones (ciprofloxacin and nalidixic acid) and tetracyclines, while resistance to erythromycin and gentamicin was comparatively low. This is most probably due to the frequent use of enrofloxacin (quinolone) and doxycycline (tetracycline) in the studied farms.
Several virulence factors have been identified in Campylobacter, which include flagella-mediated motility, bacterial adherence to intestinal mucosa, invasive capability, and the ability to produce toxins. C. jejuni isolates were positive for the presence of most of the virulence genes analyzed related to these virulence factors. However, few strains were negative for the flaA gene, while all were positive for the flaB gene. Those adjacent genes encode for the protein flagellin, composing the flagellar filament, an important colonization factor (Silva et al., 2011; Koolman et al., 2015). Isolates negative for the flaA gene might have reduced motility and colonization ability (Neal-McKinney et al., 2010). The gene wlaN, responsible for the expression of Guillain–Barré syndrome, was detected with a low frequency, in agreement with other reports (Datta et al., 2003; Talukder et al., 2008; Koolman et al., 2015). In contrast, the multidrug CmeABC efflux system, which has a role in antimicrobial resistance, was present in all but one isolate that lacked the CmeB gene. The efflux system is common in Campylobacter and consists of an external membrane protein (CmeC), a drug transporter in the internal membrane (CmeB) and an external membrane protein (CmeA). They all form a membrane channel that expels toxic substances from the cell (Lin et al., 2002). The hipO gene, which is specific for C. jejuni, was not present in any of the C. coli isolates. Besides this gene, and the wlaN gene, which is relatively rare, all virulence genes analyzed were present in all C. coli strains. It is noteworthy to highlight that the sodB gene involved in stress defense, and recently first reported in C. coli isolates by Koolman et al. (2015), has also been found in all C. coli isolates in this study.
Altogether, the in-depth characterization of these poultry isolates contributes to the understanding of Campylobacter epidemiology. WGS technology has become a fast and affordable tool and may become a rapid and cost-effective approach to characterize isolates from epidemiological studies (Llarena et al., 2017).
Footnotes
Acknowledgment
This study was partially supported by the CamCon project (Campylobacter control—novel approaches in primary poultry production), funded by the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 244547.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.