
Abstract
Shigella flexneri is a major health problem in developing countries. There are 19 serotypes recognized based on O-antigen structure and its typing is important for epidemiological purposes. However, the diversity of serotypes and the difficulties presented by phenotypic serotyping, for example, unavailable antisera for less common antigens, require the implementation of molecular techniques. In this study, we developed two multiplex PCR assays targeting the O-antigen synthesis genes and the O-antigen modification genes, for the rapid identification of S. flexneri serotypes 1/7, 2, 4, 5, and 6 (PCR A) and serotype 7 and group antigenic factors (3,4; 6; 7,8; E1037) (PCR B). A total of 73 S. flexneri strains representing 18 serotypes, except serotype 1d, were used in the study. Specific amplification patterns were obtained for each of the different serotypes. All strains tested had concordant results with phenotypic and genotypic serotyping; therefore, its implementation in the microbiology clinical laboratory will significantly improve S. flexneri serotyping.
Introduction
S
The genus Shigella comprises four species: Shigella dysenteriae, Shigella flexneri, Shigella boydii, and Shigella sonnei. S. flexneri is the prevalent species in developing countries (Kotloff et al., 1999; Boletín Integrado de Vigilancia, 2012). Except for S. sonnei, each species contains multiple serotypes based on the structure of the O-antigen.
Serotype-specific Shigella surveillance is important to monitor the burden of disease, to investigate outbreaks, to inform and evaluate programmatic activities, and to elaborate national policies aimed to reduce the incidence of shigellosis. To establish the diversity of the circulating serotypes and their distribution in time and space, it is necessary to determine the antigenic characteristics of the Shigella strains in the different regions. In fact, serotyping using rabbit antisera or monoclonal antibodies raised against specific types and group factors is considered as the gold standard typing method in epidemiological surveillance (WHO, 1999; van der Ploeg et al., 2010).
Up to now, 19 well-established S. flexneri serotypes (1a, 1b, 1c or 7a, 1d, 2a, 2b, 3a, 3b, 4a, 4av, 4b, 5a, 5b, 6, 7b, X, Xv, Y, and Yv) have been recognized (Brenner, 1984; Talukder et al., 2002; Ye et al., 2010; Sun et al., 2012; Qiu et al., 2013; Shashkov et al., 2013), but other serotypes are also possible (Knirel et al., 2015).
The O-antigen of all S. flexneri serotypes (except serotype 6) shares a linear tetrasaccharide repetition composed of one N-acetylglucosamine (GlcNAc) and three rhamnose residues (→2)-α-
The genes involved in the biosynthesis of S. flexneri O-antigens are located in the chromosome. The glucosylation and O-acetylation are mediated by a cluster of three gtr genes (gtrA, gtrB, gtr type) or a single gene (oac), respectively, which are carried by bacteriophages (Allison and Verma, 2000). gtrA and gtrB genes are highly homologous and interchangeable, whereas gtr type gene (gtrI, gtrIC, gtrII, gtrIV, gtrV, and gtrX) encodes the serotype-specific glucosyl transferase (Allison and Verma, 2000). Moreover, a plasmid-encoded gene lpt-O is responsible for the O-antigen modification that results in the addition of PEtN (Sun et al., 2012).
Except for serotypes 3, X, and Y, the other serotypes have a specific gtr gene, for example, gtrII for S. flexneri 2 (Simmons, 1971; Allison and Verma, 2000). In particular, S. flexneri 7 has the gtrI gene (specific for S. flexneri 1) in addition to the gtrIC gene (specific for S. flexneri 7) (Stagg et al., 2009).
Serotyping by traditional methods has some disadvantages: (1) it requires several different typing antisera which are expensive, (2) antisera for less common antigens are often unavailable, and (3) serotyping rough strains can be very difficult, if not impossible. For all these reasons, the development of alternative typing methods is required. Therefore, the aim of the present study was to develop two multiplex PCR assays based on DNA markers located within O-antigen modifying genes. The designed multiplexes offer a simple and economic solution for serotyping that can be applied in any microbiology clinical laboratory. The results obtained with the multiplexes designed in this study match with the traditional method providing reliable epidemiological data.
Materials and Methods
Bacterial strains
The reference strains and clinical isolates used in this study belong to the collection of Servicio Antígenos y Antisueros. All strains were biochemically characterized in accordance with the procedures previously described (Edwards and Ewing, 1986) and kept at −70°C in tryptic soy (TS) broth with glycerol.
Serotyping was performed by slide agglutination assay using specific antisera (Instituto Nacional de Producción de Biológicos (INPB), ANLIS “Dr. Carlos G. Malbrán,” Argentina) and monoclonal antibody S. flexneri MASF IV-1 (Reagensia AB, Sweden). A total of 73 S. flexneri strains (Table 1) representing 18 serotypes, except serotype 1d which is not available in our collection, were used to assess the conditions for both multiplex PCRs.
Number of strains used per serotype.
To check the specificity of the primers, we used 34 strains of all currently recognized serotypes of other Shigella species as follows: S. dysenteriae (n = 13), S. boydii (n = 19), and S. sonnei (n = 2) and 7 Escherichia coli enteroinvasive (EIEC) pathogroup strains (O28ac:NM, O29:H10, O112ac:NM, O124:H30, O136:NM, O164:NM, and O167:H5).
Preparation of DNA templates
All strains examined by PCR were grown on TS agar plates at 37°C. DNA was extracted from bacteria by resuspending a single colony in 300 μL of sterile water, boiling the suspension for 15 min, followed by centrifugation at 13,000 g for 10 min. The supernatant was then used as the DNA template for PCR.
PCR primers
Primer design and in silico analysis of cross-reactivity and hairpin formation were done using Primer3 (primer3
One forward primer, specific for gtrB gene, and 4 reverse primers were designed to amplify S. flexneri serotype-specific genes gtrI, gtrII, gtrIV, and gtrV (Fig. 1). Primer pairs for S. flexneri 6 and S. flexneri factors 3,4, 6 and 7,8 were designed to amplify the wzx, rfc, oac, and gtrX genes, respectively.
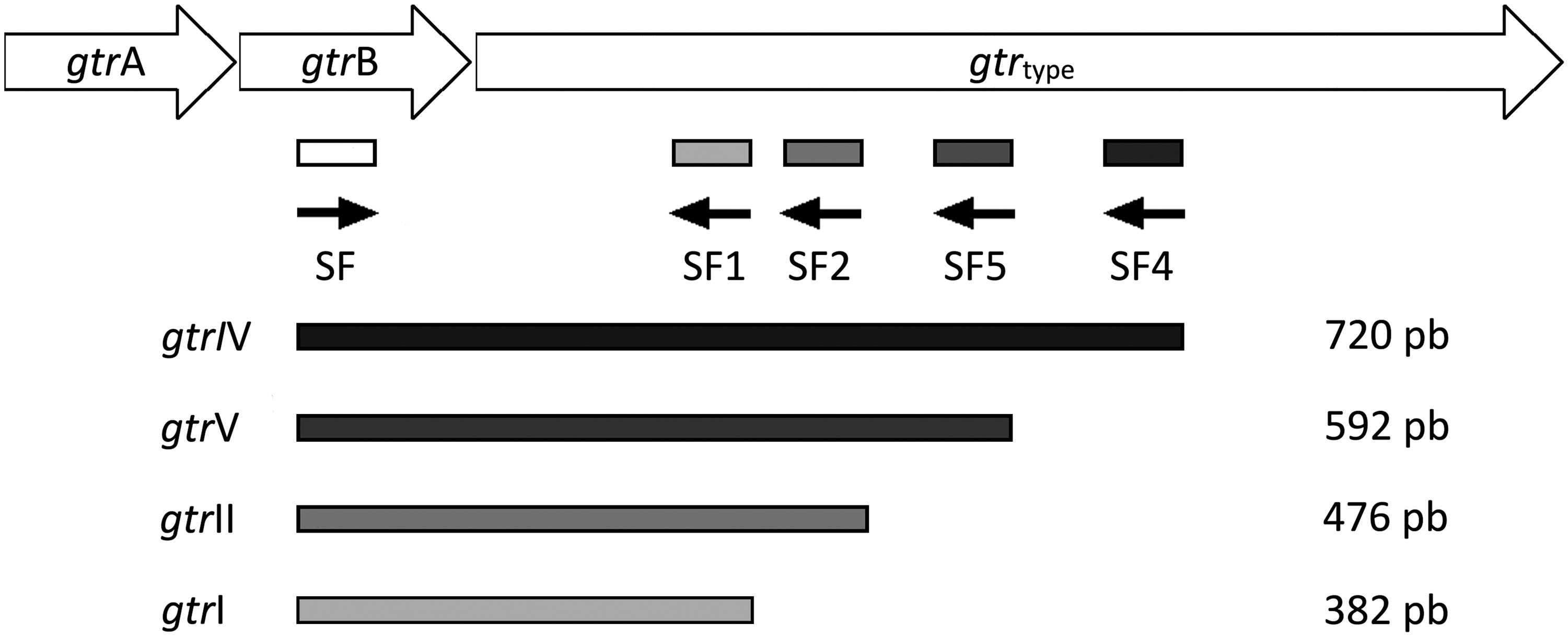
Scheme of the binding sites of the primers designed for the amplification of the 1, 2, 4, and 5 Shigella flexneri serotypes.
Primers used for the amplification of the lpt-O and gtrIC genes were taken from previous publications (Sun et al., 2011, 2012).
The primers were synthesized commercially and dissolved in ultrapure water to obtain a 100 μM stock solution. Primers are listed in Table 2.
NA, not applicable.
PCR amplification and detection
The optimal conditions defined for the assay are described below. Each multiplex PCR was carried out using a total volume of 25 μL containing 1 × PCR buffer, 1.5 mM MgCl2, 1 U Taq DNA polymerase, 200 μM dNTP, primers (SF1, SF2, and SF5: 0.125 μM; SF, SF4, SF6F, SF6R, gtrICF, gtrICR, FG3,4F, FG3,4R, FG6F, FG6R, FG7,8F, and FG7,8R: 0.25 μM; lpt-OF and lpt-OR: 0.50 μM), and 5 μL of DNA template. The primers included in the multiplex “PCR A” were: SF, SF1, SF2, SF4, SF5, SF6F, and SF6R and those included in the multiplex “PCR B” were: FG3,4F, FG3,4R, FG6F, FG6R, FG7,8F, FG7,8R, gtrICF, gtrICR, lpt-OF, and lpt-OR.
The amplification parameters were as follows: a preheat step of 94°C for 5 min, followed by 30 cycles of 94°C for 60 s, 55°C for 90 s, and 72°C for 90 s, with a final extension of 72°C for 7 min in a thermocycler (Labnet International, Inc., EEUU).
A portion (10 μL) of the reaction mixture was mixed with loading buffer, subjected to electrophoresis in 2% agarose gel, stained with ethidium bromide, and visualized by UV transillumination (DyNA Light; Labnet, EEUU).
Results
Initially, we tested the ability of each primer pair to amplify specific target DNA sequences. Amplicons of the expected sizes were obtained from all control strains, confirming the specificity of the primers (data not shown).
The reaction conditions for the multiplex PCR assays were optimized as follows to ensure that all the target gene sequences were satisfactorily amplified. We performed the multiplex PCR testing different annealing temperatures (from 51°C to 61°C) and primer concentrations (from 0.125 to 1 μM) (data not shown). The annealing temperature of 55°C was selected because at this temperature it was possible to observe with adequate resolution all the amplified products and in addition the reaction did not produce nonspecific nucleotide fragments. The amplicon sizes showed good separation on 2% agarose gels and the PCR products could be unequivocally identified by its size (Figs. 2 and 3).
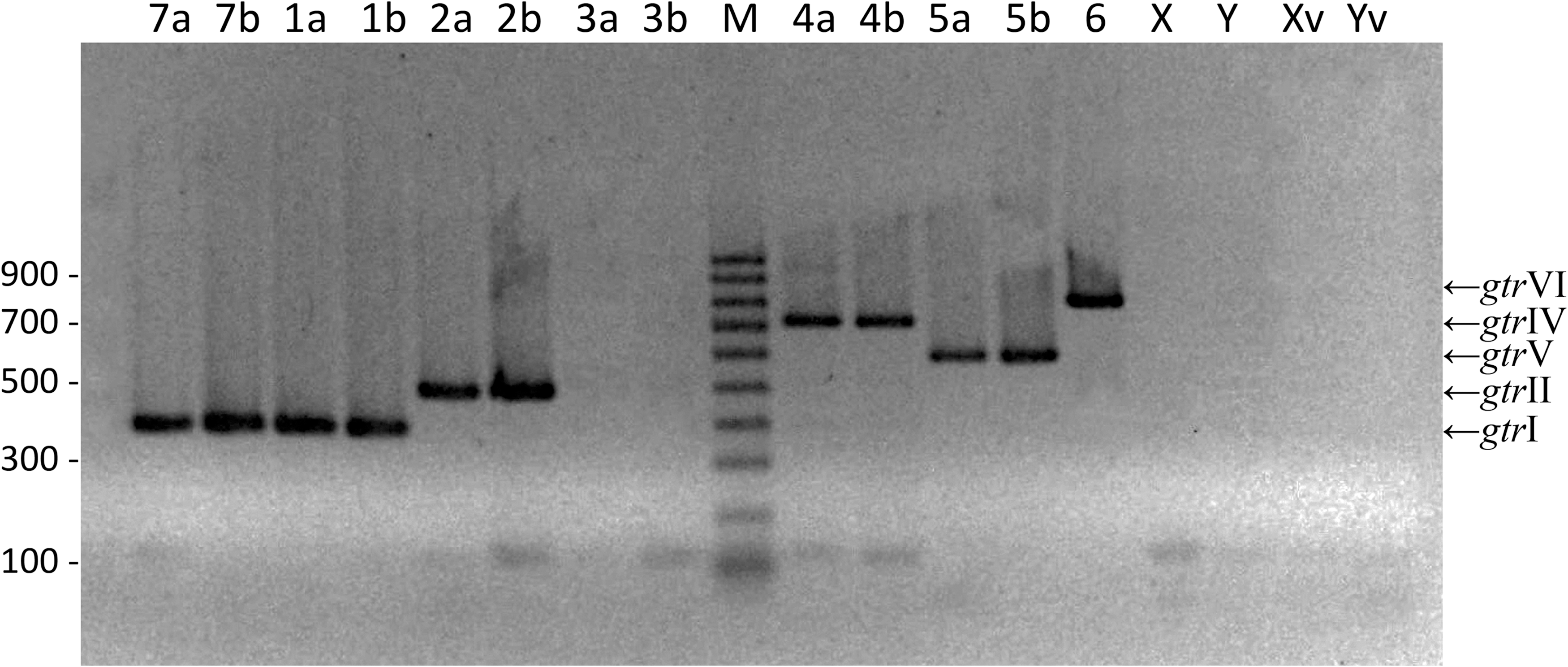
Multiplex “PCR A.” Multiplex PCR products of genes gtrI (382 pb, serotypes 7a, 7b, 1a, and 1b), gtrII (476 pb, serotypes 2a and 2b), gtrIV (720 pb, serotypes 4a and 4b), gtrV (592 pb, serotypes 5a and 5b), and wzx (829 pb, serotype 6). Serotypes are indicated in the upper region of the gel. M: DNA molecular size marker (Sigma; 100 pb ladder).
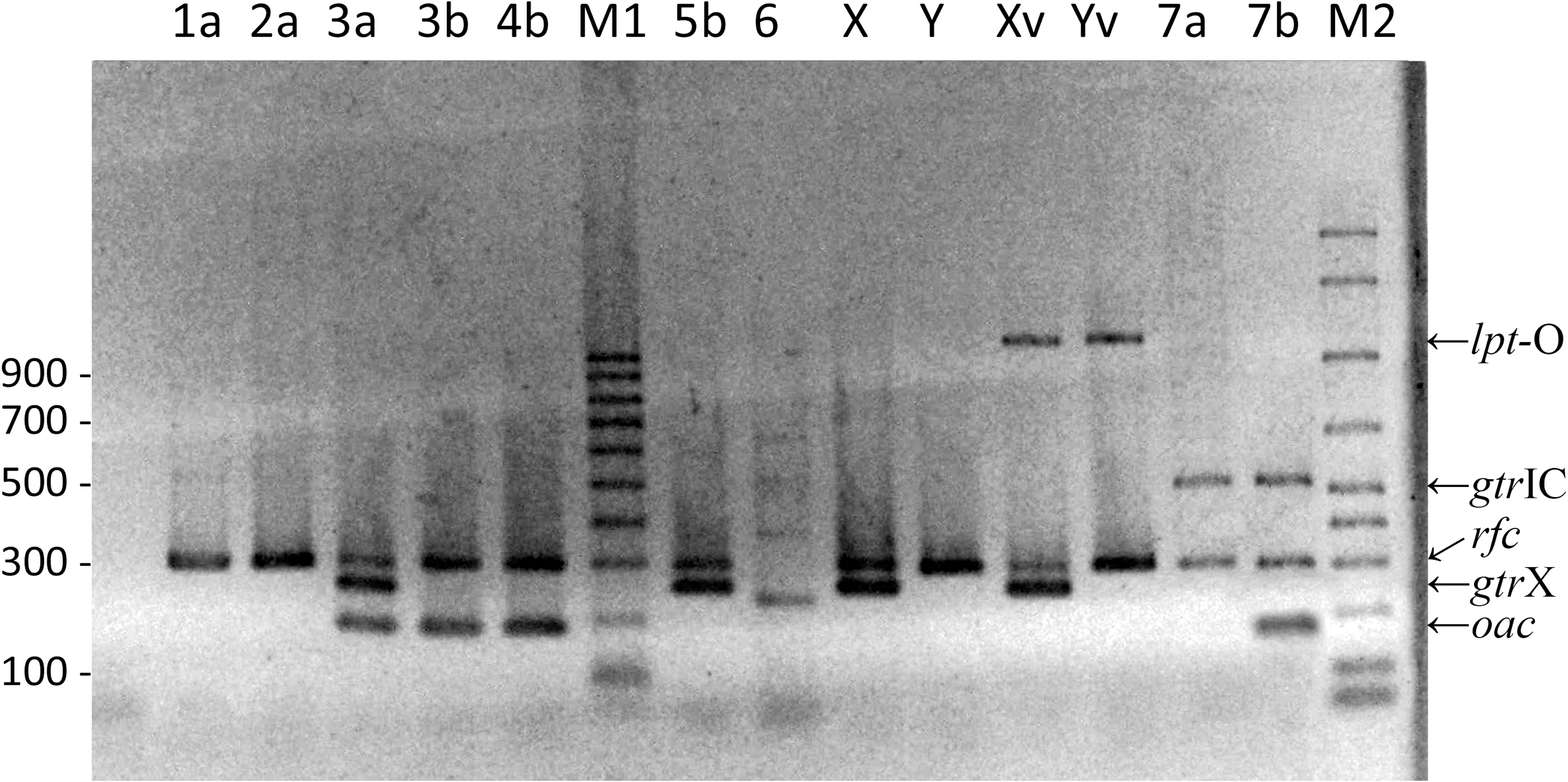
Multiplex “PCR B.” Multiplex PCR products of genes rfc (296 pb), oac (382 pb), gtrX (251 pb), lpt-O (1098 pb), and gtrIC (518 pb). Serotypes are indicated in the upper region of the gel. M1: DNA molecular size marker (Sigma; 100 pb ladder). M2: DNA molecular size marker (Bio-Rad; 50 pb ladder).
As expected, in the multiplex “PCR A” there was no amplification in the serotypes 3, X, and Y. The bands corresponding to serotypes 1, 2, 4, 5, and 6 were clearly observed as shown in Figure 2. Furthermore, the band corresponding to the amplification of the gtrI gene was observed in serotype 7 (Fig. 2).
In the multiplex “PCR B,” the rfc gene, present on the chromosome of S. flexneri, was amplified in all the strains except S. flexneri 6. The bands corresponding to the group factors were clearly observed, as well as the band corresponding to serotype 7 (Fig. 3).
Both multiplexes did not amplify any products from any strains of different Shigella species or EIEC (data not shown).
Specific amplification patterns were obtained for each of the different serotypes (Table 3). The final results were obtained by analyzing the bands of both multiplexes. For example, in PCR A we obtained amplification of gtrII gene and in PCR B we obtained amplification of rfc and gtrX genes. As a consequence, the isolate tested corresponds to serotype 2b, as shown in Table 3. Moreover, we performed the correlation between the results obtained by serotyping and those obtained by gene amplification (Table 3). All strains tested had concordant results with phenotypic and genotypic serotyping.
van der Ploeg et al. (2015).
NA, not applicable.
Discussion
S. flexneri infections are a major health problem in children, mainly in developing countries (Kotloff et al., 1999; Kirk et al., 2015). The epidemiological surveillance is an important aspect in the control of the disease, allowing us to know the diversity of the circulating serotypes and their distribution in time and space (Kotloff et al., 1999; van der Ploeg et al., 2010). Moreover, surveillance is also necessary to evaluate programmatic activities and to elaborate national policies aimed to reduce the incidence of shigellosis throughout the country.
Phenotypic serotyping is the gold standard method but it has some disadvantages. As an example of this, commercial antisera are expensive and do not cover all the possible epitopes of the O-antigen (van der Ploeg et al., 2010). For these reasons, the use of molecular biology, such as the PCR amplification, to target specific genes is an alternative typing method, although it is essential for these novel strategies to be correlated with serotyping.
In that sense, in 2011 a multiplex PCR was published by Sun et al. (2011), but that work did not include either S. flexneri serotype 6 or group antigen E1037 which are both circulating in Latin America (Boletín Integrado de Vigilancia, 2012). More recently, another group published a modification of the methodology described by Sun et al. using real-time PCR and whole-genome sequencing to investigate the phenotypic and genotypic serotype identifications (Gentle et al., 2016). Because of the complexity and costs, the latter methodologies are not applicable in most clinical microbiology laboratories of Latin America. Therefore, to have a low-cost assay, we developed two multiplex PCR protocols (“PCR A” and “PCR B”) for the O-antigens by targeting the genes encoding the immunologically recognized antigens.
The multiplex “PCR A” was designed to define serotype genes while the multiplex “PCR B” to identify group antigenic factor genes and serotype 7 specific gene (gtrIC). The combination of both assays is able to efficiently determine the 19 serotypes recognized up to now.
Our method to type S. flexneri involves an economic technique that many microbiology laboratories can easily implement: simple and fast DNA template preparation, only two multiplex PCRs to determine all S. flexneri serotypes, and easy analysis of the amplification products by agarose gel electrophoresis. Moreover, the presence of the rfc band corresponding to the amplification of the group factor 3,4 can be used as a control (except in serotype 6) because it guarantees no drawbacks in the DNA extraction process or the cycling conditions.
It is important to note that both methodologies, serotyping and multiplex PCRs, should be used as complementary techniques, since both have certain disadvantages. In terms of serology, since it is impossible to consider antigenic determinants not described at the moment, polyclonal antisera with unknown cross-reactivity could be produced leading to false positive results. As an example, a multidrug-resistance epidemic clone S. flexneri Xv isolated in China could have been typified as S. flexneri 4 by the use of commercial antisera, although these bacteria do not have the gtrIV gene (Ye et al., 2010). In terms of PCR, the presence of the gene does not guarantee its expression because deletions, insertions, and mutations in O-antigen modification genes render it inactive (Sun et al., 2011; Gentle et al., 2016).
Conclusions
The performance of the multiplexes correlates perfectly with antigenic serotyping, although future validation studies are required to be able to use it in the epidemiological surveillance of S. flexneri.
Given the relevance and complexity of S. flexneri serotyping, it is essential to agree with the international scientific community in the implementation of a standardized protocol, which includes both methodologies, to make a global contribution in terms of surveillance.
Footnotes
Acknowledgments
This work was supported by the regular federal budget of the National Ministry of Health of Argentina. The authors thank Dr. Sonia Gomez for the critical reading of the article and helpful suggestions.
Disclosure Statement
No competing financial interests exist.