
Abstract
Salmonella enterica
serovar 1,4,[5],12:i:- has emerged over the last two decades as one of the most common serovars causing human salmonellosis in Europe. It is supposed to originate from Salmonella enterica serovar Typhimurium due to antigenic and genotypic similarities between the two serovars. Due to the high level of similarity, the multilocus variable-number tandem repeat analysis (MLVA) protocol designed for Salmonella Typhimurium routine typing is commonly used also for the characterization of S. 1,4,[5],12:i. Nevertheless, the Salmonella Typhimurium-based MLVA protocol often shows poor discriminatory power for S. 1,4,[5],12:i. Indeed, only a limited number of MLVA profiles have been described for S. 1,4,[5],12:i:-. Moreover, based on the MLVA clustering, S. 1,4,[5],12:i:- is supposed to display high clonality. The aim of the present work was to assess whether the five loci of Salmonella Typhimurium investigated by MLVA are sufficiently accurate to correctly assign S. 1,4,[5],12:i:- isolates. For this purpose, 38 epidemiologically unrelated S. 1,4,[5],12:i:- were subjected to whole-genome sequencing. Isolates were selected among a collection of monophasic strains isolated in Italy from different sources over the period 2014–2016 and belonging to the five most commonly detected MLVA profiles.
Results confirmed the possible clonality for S. 1,4,[5],12:i:- serovar in the light of the scarce difference observed in terms of single-nucleotide polymorphisms (SNPs) among investigated isolates. Nevertheless, unrelated isolates on the basis of the difference of SNP number were characterized as indistinguishable by MLVA profile, thus suggesting an insufficient resolution of MLVA. Hence, we can conclude that MLVA-based approach does not seem a valuable proxy to deepen into the epidemiological relationship among S. 1,4,[5],12:i:- isolates. These evidences can be useful to avoid incorrect assignment especially when surveillance data are used for outbreak investigations.
Introduction
In the last two decades, a new Salmonella serovar, the monophasic variant of Salmonella enterica serovar Typhimurium with antigenic formula 1,4,[5],12:i:-, became one of the three most common serovars causing human salmonellosis in Europe (Mastrorilli et al., 2018). Given the high level of similarity with Salmonella Typhimurium, which is considered its ancestor, it is commonly accepted to use the multilocus variable-number tandem repeat analysis (MLVA) protocol, developed for Salmonella Typhimurium, also to define epidemiological relationship among isolates of its monophasic variant even if this technique requires serovar-specific protocols. However, despite being highly similar to Salmonella Typhimurium, S. 1,4,[5],12:i:- presents a limited heterogeneity of MLVA profiles (Barco et al., 2015). This work aimed at investigating the possible high clonality of S. 1,4,[5],12:i:- with whole-genome sequencing (WGS), and assessing the accuracy of the five loci of Salmonella Typhimurium MLVA EU protocol (Lindstedt et al., 2004) to correctly assign S. 1,4,[5],12:i:- isolates.
Materials and Methods
Sample selection
The OIE and Italian National Reference Laboratory for Salmonella collected a number of 500 S.
Phage typing
S. 4,[5],12:i:- isolates were phage typed using the protocol (Anderson et al., 1977) and following the interpretative guidelines set out for Salmonella Typhimurium by the International Federation for Enteric Phage Typing (IFEPT, Laboratory of Enteric Pathogens, Health Protection Agency, Colindale, London, United Kingdom).
Multilocus variable-number tandem repeat analysis
MLVA was performed according to the protocol described by Lindstedt et al. (2004). The size measurements for each locus were estimated using a Genetic Analyzer 3130XL (Applied Biosystems, Life Technologies Corporation, Carlsbad, CA). According to the nomenclature suggested by Larsson et al. (2013), the MLVA results were reported as a string of five numbers representing the variable number of tandem repeats at the corresponding loci (STTR9–STTR5–STTR6–STTR10pl–STTR3), or as “–” in the case that a polymerase chain reaction (PCR) product was not obtained for a locus.
Selected isolates were distributed among the five most frequent MLVA profiles (i.e., 3-13-10-NA-0211, 3-11-9-NA-0211, 3-10-9-NA-0211, 3-11-11-NA-0211, and 3-12-9-NA-0211), representing about 35% of the entire collection and isolated in 2015–2016.
WGS and data analysis
Genomic DNA (gDNA) was extracted for WGS starting from overnight cultures plated on Agar Tryptose (AT), using the column-based QIAamp DNA Mini kit (Qiagen), following the manufacturer's instructions. Libraries for sequencing were prepared with the Nextera XT DNA sample preparation kit (Illumina): briefly, gDNA was subjected to tagmentation, a step in which the DNA is enzymatically cleaved and then tagged with Illumina adapters. Cleaved gDNA was subjected to a second PCR step for indexing with a unique combination of i5 and i7 index primers. High-throughput, paired-end sequencing (2 × 250 bp) was performed on the MiSeq sequencing platform using the v3 Reagent kit.
Resulting raw reads were assembled in contigs with Assembler 1.2 (Larsen et al., 2012) and used together with the reference genome of S. 1,4,[5],12:i:-, namely LN999997 (Petrovska et al., 2016), to build a single-nucleotide polymorphism (SNP)-based phylogenetic tree with CSIPhylogeny 1.4 (Kaas et al., 2014), using default parameters for SNP filtering and SNP pruning. All the full-length sequences have been deposited in ENA (
Results
The phylogenetic tree built using the representative European clone of S. 1,4,[5],12:i:-, isolate LN999997, as the reference genome (Fig. 1), presents S. 1,4,[5],12:i:- isolates whose SNP distance ranged from 1 to 188 SNPs. Moreover, SNP-based phylogeny showed three main clusters (Fig. 1). Irrespective of their MLVA profiles, S. 1,4,[5],12:i:- isolates were scattered into three clusters. Cluster 1 (highlighted in green) included 11 isolates, belonging to four different MLVA profiles (3-11-9-NA-211, 3-10-9-NA-211, 3-11-11-NA-211, and 3-13-10-NA-211), with an average SNP difference of 22 SNPs. The second cluster, highlighted in red, grouped together 14 isolates, falling into four different MLVA profiles (3-12-9-NA-211, 3-10-9-NA-211, 3-11-11-NA-211, and 3-13-10-NA-211), and characterized by 80 SNPs of difference. Finally, cluster 3 (highlighted in blue) consisted of 13 S. 1,4,[5],12:i:- isolates belonging to three MLVA profiles (3-11-9-NA-211, 3-11-11-NA-211, and 3-13-10-NA-211) and an average SNP difference of 30 SNPs. This latter cluster also included the reference genome. Isolates belonging to MLVA profiles 3-11-11-NA-211 and 3-13-10-NA-211 were scattered into all the three clusters, isolates belonging to MLVA profiles 3-11-9-NA-211 and 3-10-9-NA-211 were distributed between two different clusters, and all the isolates showing the single MLVA profile 3-12-9-NA-211 were assigned to a single cluster.
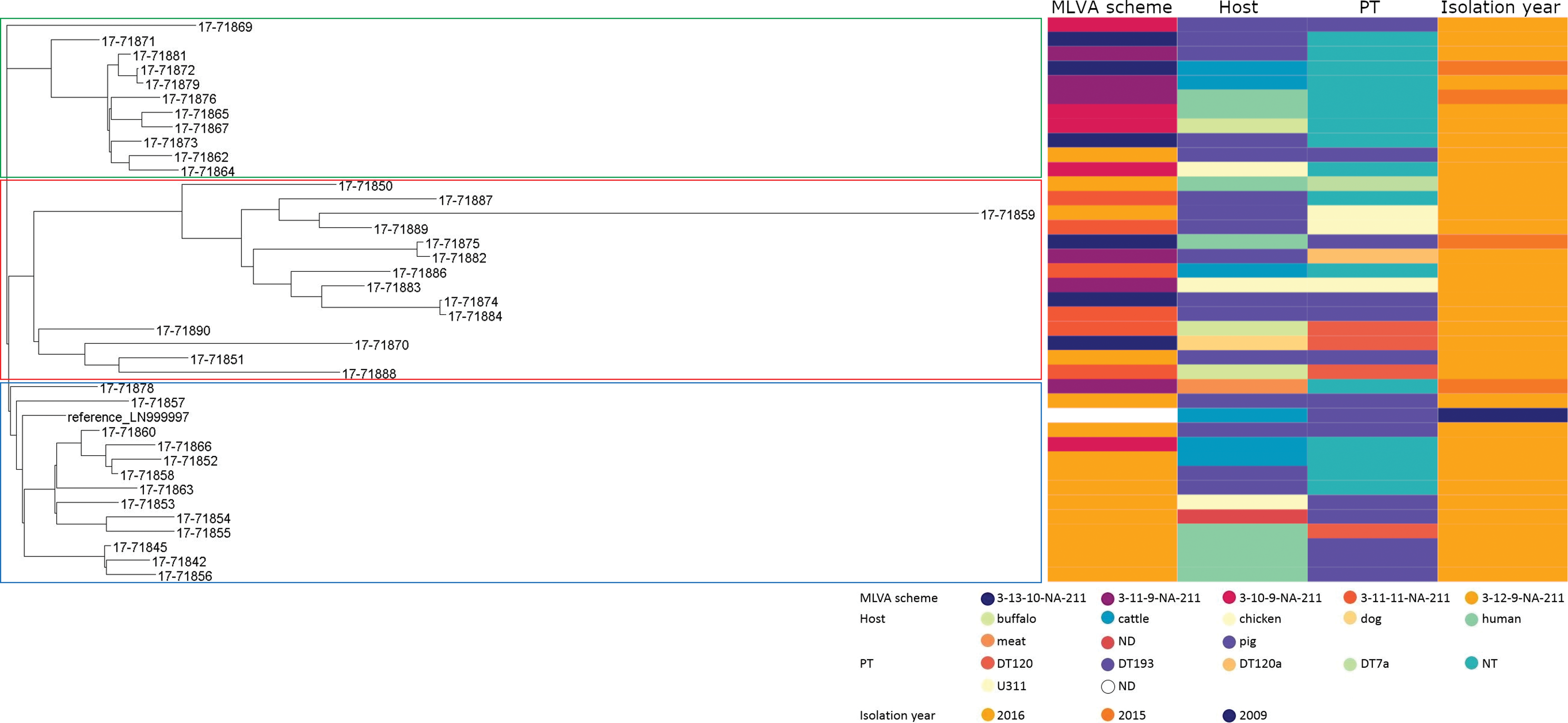
Phylogenetic tree, MLVA profile, and metadata (Host = isolation source, PT = phage type, and isolation year) for analyzed samples and reference genome LN999997. Strain clustering is highlighted (green: cluster 1, red: cluster 2, blue: Cluster 3). MLVA, multilocus variable-number tandem repeat analysis. Color images are available online.
Phage type and host for selected isolates are also shown in Figure 1.
Discussion
Despite being highly similar to Salmonella Typhimurium, S. 1,4,[5],12:i:- presents distinct epidemiological, phenotypic, and genotypic features. In particular, this serovar shows a limited heterogeneity in terms of MLVA profiles, with only two of the five proposed loci resulting polymorphic and hence discriminative in S. 1,4,[5],12:i:- characterization (Barco et al., 2015). Nevertheless, given the high reproducibility, MLVA has long been used as a subtyping method also for monophasic variant of Salmonella Typhimurium in molecular epidemiology (EFSA, 2013).
Our results likewise point out that MLVA subtyping of monophasic variant of Salmonella Typhimurium isolates seems to be scarcely discriminative. This becomes of particular concern in the case of clonal serovars such as S. 1,4,[5],12:i:-: in our analysis, indeed, a limited difference in terms of SNPs is shown (maximum pairwise SNP difference among isolates is 188 SNPs).
Despite the clonal nature of this serovar, a certain difference in terms of SNPs is clear in cluster 1 (22 SNPs) and in cluster 3 (30 SNPs), however, many of the isolates belonging to such clusters resulted indistinguishable by MLVA profile.
Epidemiological data indeed confirmed that selected strains were unrelated, supporting the inadequate resolution power of MLVA when applied in discriminating S. 1,4,[5],12:i:- isolates.
Indeed, it has already been shown that MLVA discriminatory power for S. 1,4,[5],12:i:- serovar is associated with STTR5 and STTR6 loci, with STTR10 being almost constantly absent and STTR3 and STTR9 being highly stable (Barco et al., 2015).
Since only five loci are evaluated, they could not reflect genome-wide information precisely, and thus, MLVA might not be optimal for phylogenetic analysis, especially when reduced polymorphism in the selected loci has already been observed, as in the case of S. 1,4,[5],12:i:-, which is also characterized by a clonal behavior.
However, even though a general SNP threshold could not be established to define a Salmonella outbreak, and even if the presence of tens of SNPs commonly indicates that bacteria are genetically similar and recently originated from the same source (Cibin et al., 2019 in press), epidemiological considerations and traceback information are critical to interpret WGS analyses (Pightling et al., 2018).
Current routine typing methods used for Salmonella characterization, including MLVA, showed a doubtful resolution (Morganti et al., 2018), while the greater discriminative power of WGS could likely be helpful especially with clonal pathogens, for which the MLVA-based approach does not seem a valuable typing method to deepen into an epidemiological relationship.
Conclusions
The collected data, based on the characterization of the S. 1,4,[5],12:i:- isolates obtained over a 3-year surveillance at the national level and showing the most common MLVA profiles, confirmed that this serovar can be considered clonal, being the epidemiologically unrelated investigated strains very close in terms of SNP difference. MLVA showed insufficient resolution in the characterization of such isolates, given that isolates with considerable diversity in terms of SNPs were indistinguishable by MLVA profile.
The MLVA-based approach thus does not seem a valuable proxy to deepen into the epidemiological relationship among S. 1,4,[5],12:i:- isolates. These evidences can be useful to avoid incorrect assignment especially when surveillance data are used in the context of outbreak investigations.
Footnotes
Acknowledgment
This work was supported by the Italian Ministry of Health (grant no. IZSVE RC10/14).
Disclosure Statement
No competing financial interests exist.