
Abstract
Vibrio parahaemolyticus is a common inhabitant of coastal estuaries, and can accumulate to high levels in the shellfish that populate those waters. Human gastrointestinal infection occasionally follows ingestion of raw oysters, and it can lead to extended closures of implicated oyster beds with serious economic consequences. To track down the source of human infection, and to monitor strain variation in the environment, a user-friendly and affordable typing method that provides sufficient resolution for epidemiological analysis is needed. Polymorphic locus sequence typing (PLST) is based on conventional PCR and dideoxynucleotide sequencing of the one or two most phylogenetically informative genomic loci. Bioinformatic analyses of GenBank databases identified the V. parahaemolyticus polymorphic tandem repeat-containing loci VpMT1 and VpMT2 on chromosomes 1 and 2, respectively, as promising PLST targets, yielding diversity indexes of 0.99. Phylogenetic analysis identified multiple clusters representing strains known or likely to be epidemiologically related. Correlations with serotype and multilocus sequence type were strong but resolution was higher; for example, North American ST36 strains yielded 16 VpMT1 alleles. In the laboratory, VpMT1 and VpMT2 were robust, resolving 16 of 17 strains following PCR and sequencing directly from heat-killed colonies. Finally, 4 of 13 retail oyster enrichments yielded VpMT sequences that were unique but closely related to previously characterized clinical or environmental V. parahaemolyticus isolates.
Introduction
Vibrio parahaemolyticus is a motile, Gram-negative bacterium commonly found in coastal, estuarine waters worldwide. It is also the leading cause of seafood-borne bacterial disease. A 2005 study estimated 3000 U.S. infections per year (FDA, 2005), but there has been a surge in recent years, illustrated by a roughly 10-fold increase in a northeastern U.S. region (Urquhart et al., 2016). Infection is most commonly associated with ingestion of contaminated raw or undercooked oysters or other shellfish, and is characterized by explosive, watery diarrhea along with nausea and vomiting, which generally resolve within a few days. Incidence peaks in warmer months, which correlates with seasonal increases in V. parahaemolyticus numbers in seawater and shellfish.
Clinical isolates of V. parahaemolyticus can typically be distinguished from environmental isolates by the presence of one or both hemolysin genes tdh and trh, although their absence in up to 28% of clinical isolates clearly indicates that other virulence mechanisms exist (Jones et al., 2012; Banerjee et al., 2015; Haendiges et al., 2015; Raghunath, 2015; Xu et al., 2015); primary candidates are the T3SS2 type III secretion system and associated Vop effectors (Ham and Orth, 2012). With respect to serotype, an O3:K6 strain emerged in India in 1996 and subsequently spread worldwide to become a pandemic clonal complex (Ceccarelli et al., 2013; Espejo et al., 2017). O and K antigen typing has not, however, been particularly useful in tracking this spread, since strains within this complex have demonstrated 27 serovariants of the original O3:K6 (Espejo et al., 2017). More recently, a particularly virulent strain with serotype O4:K12, indigenous to the U.S. Pacific Northwest, has spread to coastal regions on both sides of the Atlantic where it has been responsible for multiple outbreaks (Martinez-Urtaza et al., 2013, 2017).
As an alternative to serotyping, multiple DNA-based typing methods have been developed and used for evolutionary and epidemiological analysis of V. parahaemolyticus (Espejo et al., 2017). Pulsed-field gel electrophoresis (PFGE) was long considered the gold standard and provides high level strain resolution (Haendiges et al., 2015), but the method is technically complex and time consuming. Multilocus sequence typing (MLST), examining single nucleotide polymorphisms (SNPs) in seven relatively conserved housekeeping genes, has provided the most reliable and informative data. For example, it was used to demonstrate the recent spread of sequence type 36 (ST36) strains (strongly correlated with serotype O4:K12 noted above) from the Pacific Northwest to the Atlantic Northeast, where they represented nearly 50% of clinical isolates from the New England states (Xu et al., 2015). With respect to ST36, the study also showed clear congruency between MLST and whole genome sequencing, which enhances strain resolution by extending MLST analysis to SNPs throughout the core genome (cgMLST) (Haendiges et al., 2016; Gonzalez-Escalona et al., 2017). As emphasized by Urquhart et al. (2016), strain typing on a regional level represents the best approach to assessing the risk associated with shellfish consumption, rather than the current approach that is limited to tdh/trh detection. However, since MLST and cgMLST are costly, and length-based methods such as PFGE and multilocus variable-number tandem-repeat analysis (MLVA; Kimura et al., 2008; Harth-Chu et al., 2009; Jiang et al., 2016) have intrinsically limited data portability (i.e., ability to unambiguously compare data from lab to lab and day to day), alternative sequence-based typing methods warrant exploration.
In contrast to MLST and cgMLST, but in common with MLVA, polymorphic locus sequence typing (PLST) targets tandem repeat-containing loci. Also known as microsatellites, tandem repeats range from simple dinucleotide repeats to complex CRISPR elements. Due to slippage during DNA replication, tandem repeats undergo insertion/deletion (indel) events at relatively high frequencies (Zhou et al., 2014). In MLVA, electrophoresis is used to determine locus length and hence variation in repeat number. However, tandem repeats can be imperfect, and DNA slippage events can be inexact, resulting in indels that are fractions of a repeat unit and in the accumulation of SNPs. Also, since tandem repeat-containing loci are intrinsically less conserved, indels and SNPs may extend to the regions flanking the tandem repeat. Consequently, sequence-based PLST analysis of only one or two carefully selected loci can provide, at lower cost and more rapid turnaround time, strain resolution exceeding MLST or MLVA, and approaching cgMLST. Recent studies have described the development and evaluation of PLST typing services for the foodborne pathogens Listeria monocytogenes and Salmonella enterica (Edlind and Liu, 2015; Edlind et al., 2017). These studies have also demonstrated that PLST, based on conventional PCR and dideoxynucleotide sequencing, is technically robust and hence can be used to type pathogens directly from food enrichments and other crude samples. Here, we extend PLST to V. parahaemolyticus, and demonstrate enhanced resolution relative to MLST and correlation with epidemiological data.
Materials and Methods
Bioinformatics
Candidate PLST loci were identified using Tandem Repeats Database (
Strains and lysate preparation
From frozen glycerol stocks, 17 V. parahaemolyticus strains (Table 1) were streaked for isolation on marine agar (Baltimore Biological Laboratory, Sparks, MD) with incubation at 37°C overnight. Single colonies were suspended in 200 μL Tris/EDTA (10/1 mM, pH 8.0), and heat-killed lysates were prepared by boiling in a water bath for 10 min. To test loci stability, single colonies from four different strains were streaked to fresh plates and incubated at 37°C overnight. This passaging was repeated daily for 10 d, and colonies from the first and final plates were used to prepare lysates as described above.
Vibrio parahaemolyticus Strains Used in This Study
Serotype: NA, information not available; KUT, K untypable.
Source: ATCC, American Type Culture Collection, Manassas, VA; FDA, U.S. Food and Drug Administration, Gulf Coast Seafood Laboratory, Dauphin Island, AL; USDA, U.S. Department of Agriculture, Agricultural Research Service, Dover, DE; Univ. DE, University of Delaware, Department of Biological Sciences, Newark, DE.
PCR and sequencing
Colony lysates were centrifuged (14,000 × g for 2 min) to pellet debris, and 1 μL aliquots were used as templates in 20 μL PCR mixtures with Taq polymerase as recommended by the manufacturer (New England Biolabs, Ipswich, MA). Primers (Table 2) were designed based on conserved sequences flanking the tandem repeats and synthesized by IDT (Coralville, IA). Amplification was for 32 cycles of 94°C 20 s, 55°C 1 min, and 70°C 1 min; final extension was 70°C 3 min. PCR products (5 μL) were analyzed by 1% agarose gel electrophoresis and visualized with blue light illumination after staining with SYBR Safe (Invitrogen, Grand Island, NY). For DNA sequencing, PCR products (1–3 μL) were treated with ExoSAP-IT as recommended by the manufacturer (Affymetrix, Santa Clara, CA), sequencing primer was added to 2 μM, and samples were submitted to GENEWIZ (South Plainfield, NJ). DNA sequences were edited as needed based on visual inspection of the chromatograms and trimmed to common termini.
DNA Primers Used in This Study
Typing from oyster enrichments
Retail oysters harvested from four different locations along the eastern U.S. coast were purchased in mid-September 2018. After shucking, oysters were individually macerated and 50 μL used to inoculate 5 mL of alkaline peptone water (APW) medium. Following incubation at 35°C for 20 h, 1 mL was transferred to microfuge tubes and lysates prepared by centrifugation, suspension in Tris/EDTA, and heating as described above for colonies. DNA was purified from lysates on silica columns (EPOCH Life Science, Sugar Land, TX) as recommended by the manufacturer. PCR with VpMT primers and sequencing were as described above. Aliquots of the same APW enrichments were also streaked for isolation on thiosulfate citrate bile salts sucrose (TCBS) agar (Hardy Diagnostics, Santa Maria, CA), with incubation at 35°C for 20 h. Lysates of blue-green colonies were prepared, DNA purified, and subjected to PCR and sequence analysis as described above.
Accession numbers
GB accession numbers for the VpMT1 and VpMT2 nucleotide sequences determined in this study are MH464546–MH464583 and MK309931–MK309940.
Results
Identification of candidate PLST loci VpMT1 and VpMT2
Tandem repeats were bioinformatically identified in the full-length sequences for chromosomes 1 and 2 of strains FDA_R31 and CDC_K4557, representing oyster and clinical isolates, respectively, from Louisiana (Ludeke et al., 2015). Tandem repeats plus 500 nucleotide flanking sequences were used as queries in BLASTN searches of the 23 V. parahaemolyticus genome sequences in the GB Nucleotide/nr database to identify loci that combine (1) high polymorphism, (2) presence in all V. parahaemolyticus strains, (3) lack of insertion sequences or genomic rearrangements, (4) conserved sequences within the flanks for primer design, and (5) length between 500 and 900 bases to facilitate amplification and sequencing. These screens identified loci VpMT1 and VpMT2 on chromosomes 1 and 2, respectively, as the most promising PLST candidates. Clustal alignments of the tandem repeat-containing regions of these loci from representative strains are shown in Figure 1. The VpMT1 repeat unit (TCTCTA; within locus tag VP2892 of strain RIMD2210633) encodes (on the complementary strand) a Glu-Ile repeat within a hypothetical protein, while the VpMT2 repeat unit (CAACAG; within locus tag VPA1455) encodes a Gln repeat, also within a hypothetical protein. Polymorphism is evident in the form of both indels of the tandem repeats and single or multiple nucleotide variants in the flanking regions. A literature search revealed that VpMT1 and VpMT2 both include variable number tandem repeat (VNTR) components of published MLVA studies (Kimura et al., 2008; Harth-Chu et al., 2009; Ansede-Bermejo et al., 2010; Jiang et al., 2016) with diversity indexes (DI) = 0.88–0.92 and 0.90–0.96, respectively, the latter representing the VNTR yielding the highest DI and most alleles.

Alignment of VpMT1 and VpMT2 sequences from Vibrio parahaemolyticus strains representing multilocus sequence typing (MLST) types most commonly associated with clinical disease in N. America: CFSAN007449 (ST3), 10329 (ST36), and VP2007-095 (ST631). Tandem repeats are indicated in lowercase, indels are represented by dashes, variations in flanking regions are in bold, and asterisks represent conserved sequence.
VpMT1 and VpMT2 loci were subsequently used as queries in BLASTN searches of the GB Refseq genomes database. This database includes >700 V. parahaemolyticus strains; the analysis here focused on the ca. 330 strains isolated from North American clinical isolates, oysters, or coastal waters. Clustal alignments and phylogenetic analyses of VpMT1 and VpMT2 sequences extracted from these genomes resolved these strains into 161 and 125 alleles, respectively (Supplementary Fig. S1). (The lower number of VpMT2 alleles is due to a higher rate of incomplete sequence assembly across its longer repeat.) From these data, the DI for both VpMT1 and VpMT2 were determined to be 0.99. This improvement in VpMT sequence-based strain resolution relative to the VNTR length-based resolution noted above results from polymorphism on the sequence level of both tandem repeat and flanking sequence. Additionally, BLASTN analyses indicated that both VpMT1 and VpMT2 were conserved in related Vibrio spp. including V. antiquarius, V. campbellii, and V. harveyi but absent or highly diverged in more distantly related Vibrio spp. V. vulnificus and V. cholerae and in all non-vibrio species represented in GB databases.
Laboratory evaluation of VpMT1 and VpMT2 with colony lysates
Isolated colonies from 17 V. parahaemolyticus strains (Table 1) were suspended in buffer and heat inactivated. Following centrifugation, the supernatants were used as templates for PCR with VpMT1 and VpMT2-specific primers. All 17 strains yielded single distinct products (an additional strain that failed to yield either product was subsequently shown by 16S rRNA gene amplification and sequencing to be V. vulnificus). Products were sequenced and phylogenetically analyzed along with corresponding sequences from selected GB strains to generate dendrograms, as shown in Figure 2. VpMT1 and VpMT2 each resolved 16 of the 17 V. parahaemolyticus laboratory strains. Note that one strain pair (SPRC_10290 and CPA_7081699) not resolved by VpMT1 was resolved by VpMT2, and similarly a second pair (ATCC_17802 and ATCC_17803) not resolved by VpMT2 was resolved by VpMT1, such that their combination resolved all 17 strains.
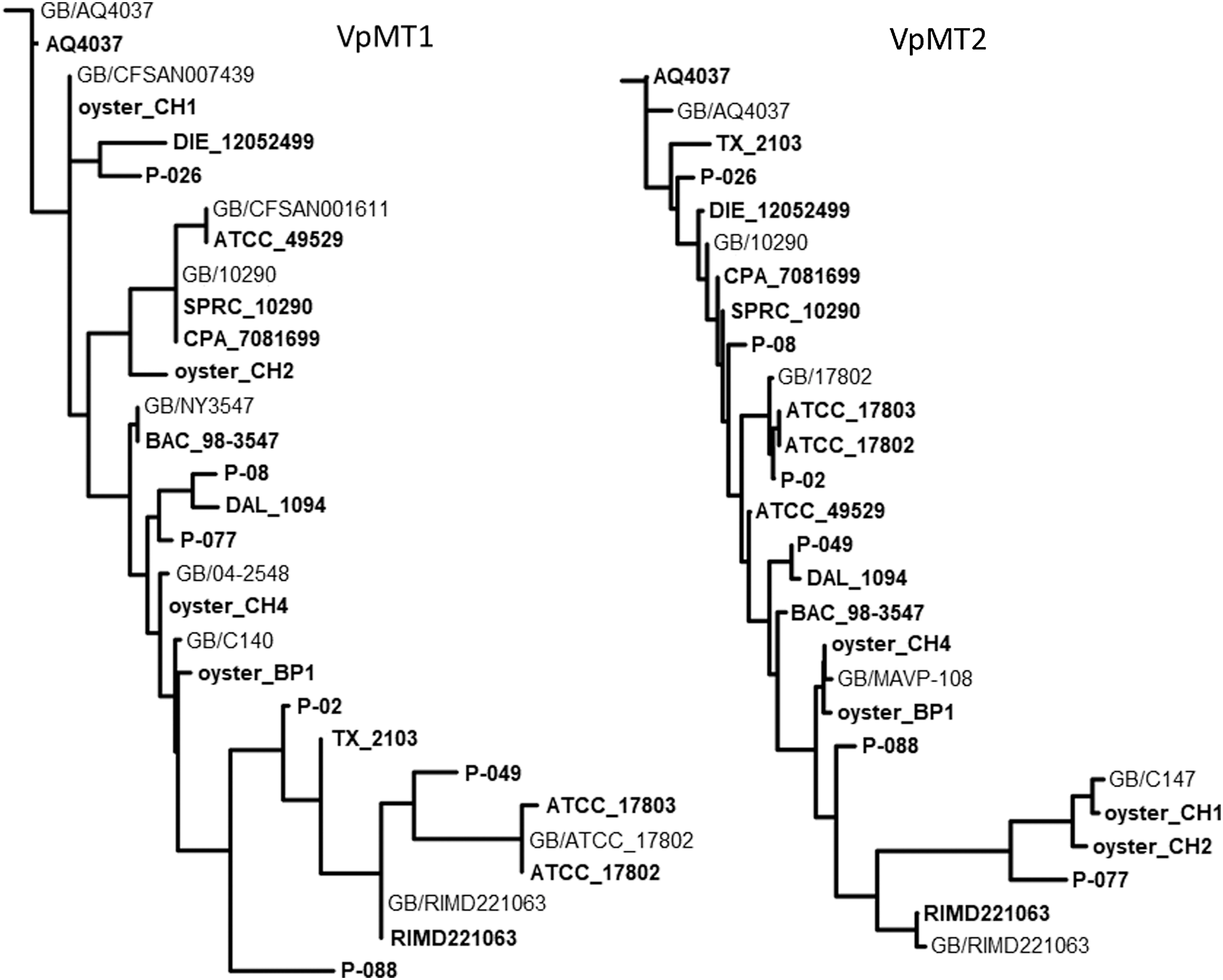
Dendrograms of VpMT1 and VpMT2 sequences amplified from heat-inactivated colonies of Vibrio parahaemolyticus laboratory strains, or directly from oyster enrichments. Selected sequences from related GenBank strains (GB/) were included for comparison.
VpMT1 and VpMT2 loci stabilities were tested by passaging four different strains by spreading on fresh plates daily for a total of 10 passages. Cells from the first and final plates were used to prepare lysates as described above. Following amplification and sequencing, no differences were observed for VpMT1; however, indels in the VpMT2 tandem repeat were observed with three of the four strains. This relative instability is consistent with the higher diversity index associated with the VpMT2-associated VNTR in MLVA studies as noted above.
As expected, comparisons between laboratory-derived and GB-derived VpMT1 sequences from the same (or likely the same) strain indicated identity for four such pairs; specifically, RIMD2201633 and GB/RIMD2201633, BAC_98-3547 and GB/NY3547, SPRC_10290 and GB/SBR_10290, and ATCC_17802 and GB/ATCC_17802 (Fig. 2). A likely addition to this list is TX_2103 and unassembled short read sequences for this same strain in the GB SRA database, while strain pair AQ4037 and GB/AQ4037 exhibited a minor difference (1 repeat unit indel) in VpMT1 sequence. Relatedly, these same strain pairs yielded sequences for the less stable VpMT2 locus that were closely related but not identical (Fig. 2; note that an assembled VpMT2 locus for GB/NY3547 was lacking).
VpMT typing directly from oyster enrichments
As a PCR-based method, PLST has demonstrated compatibility with crude samples including ground beef and poultry enrichments (Edlind et al., 2017). Thirteen retail oysters harvested from four different locations on the eastern U.S. coast were purchased in mid-September, and individually used to generate enrichment cultures in APW, the medium specified in FDA laboratory guidelines (Kaysner and DePaola, 2004). Bacterial cell pellets were suspended in buffer, lysed by heating, and their DNAs purified. Following PCR with VpMT1 primers and gel electrophoresis, products were detected with DNAs from one to three oysters from three of the four locations (six total). Sequence analysis of the products from four of these oysters (CH1, CH2, CH4, and BP1) revealed identity or close matches to VpMT1 loci of distinct V. parahaemolyticus strains represented in GB (Fig. 2). Products from the remaining two oysters (BC1 and BP2) closely matched VpMT1 loci from GB V. campbelli and V. harveyi strains. PCR was similarly conducted with VpMT2 primers, which again yielded products from the CH1, CH2, CH4, and BP1 enrichments whose sequences closely matched corresponding loci of distinct V. parahaemolyticus database strains (Fig. 2). No products were obtained with the BC1 and BP2 enrichments, consistent with the multiple mismatches between V. campbelli and V. harveyii database sequences and VpMT2 primers (not shown).
As per FDA guidelines (Kaysner and DePaola, 2004), the APW enrichments were used to inoculate TCBS plates. Blue-green colonies characteristic of V. parahaemolyticus were detected only from the BP1 oyster enrichment. VpMT1 and VpMT2 analyses showed the expected identities of the sequences from the BP1 enrichment and its TCBS colony.
Correlations of VpMT1 PLST with epidemiological data, serotyping, and MLST
Support for the application of VpMT1 PLST to strain surveillance and epidemiological investigation is provided by the analysis shown in Figure 3, focusing on VpMT1 alleles that include multiple strains from GB databases. Specifically, the majority of strains sharing the same allele are known or likely to be epidemiologically related, based on their locations and dates of isolation; for example, the three clinical isolates from Connecticut in 2013 (CTVP27C–CTVP34C) and the four oyster isolates from Maryland in 2010 (CFSAN012491–CFSAN012494). Similarly, all 12 alleles that include serotyped strains exhibit a consistent serotype; for example, the 5 O4:KII-ambiguous (Ronholm et al., 2015) clinical isolates from British Columbia (10-4242–10-4274) and 3 O1:K20 oyster isolates from South Carolina (GCSL_R136–GCSL_R138).

Dendrogram of VpMT1 sequences from Vibrio parahaemolyticus strains known or likely to be epidemiologically related. Sequences were extracted from GenBank databases, along with strain data (isolation date, U.S. state or Canadian province source, clinical or oyster isolate, serotype, and sequence type, if known; brackets indicate strain data differences that remain consistent with epidemiological relatedness). *O4:KII strains that share ambiguous (weak) K serotype (Ronholm et al., 2015).
With respect to MLST, the most prevalent sequence type in North American clinical isolates is ST36 (Xu et al., 2015). As shown in Figure 4, VpMT1 resolved the 159 North American ST36 strains represented in GB databases into 16 alleles. Similarly, VpMT1 PLST provided enhanced resolution of two additional MLST types commonly identified in North America: ST3 (8 alleles from 15 strains) and ST631 (9 alleles from 30 strains). Also illustrated in Figure 4 is the distinction between VpMT1 phylogenetic analysis by dnaml (maximum likelihood) in which resolution is focused on SNPs, and dnapars (parsimony) in which indels are weighted along with SNPs. The former yields clusters that correlate with MLST sequence type, while the latter most clearly resolves strains within each cluster.
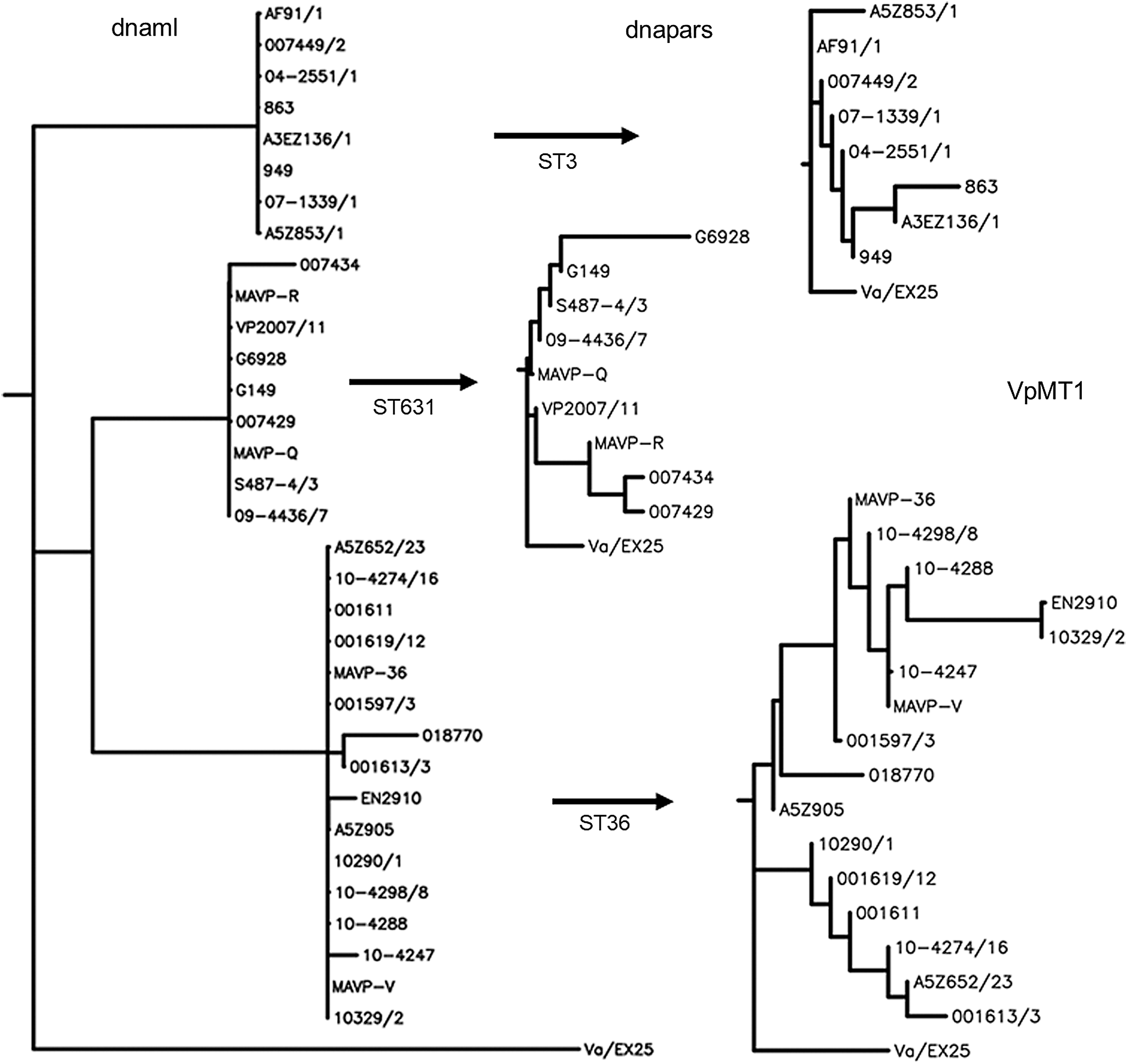
Dendrogram of VpMT1 sequences from three Vibrio parahaemolyticus multilocus sequence typing sequence types (ST3, ST36, and ST631) most commonly associated with clinical disease in N. America. dnaml, maximum likelihood analysis demonstrating resolution of strains into ST-specific clusters; dnapars, parsimony analysis demonstrating resolution of each ST (individually aligned) into multiple VpMT1 alleles. Alleles with multiple strains are indicated by strain/#, where # represents the number of additional strains. Dendrograms were rooted to the orthologous sequence from V. antiquarius strain EX25.
Discussion
V. parahaemolyticus isolates have traditionally been typed by antigen-based serotyping, and the use of strain terminology such as O3:K6 continues. However, serotyping requires costly, specialized reagents, and can yield ambiguous results (Ronholm et al., 2015); more importantly, it provides limited strain resolution. PFGE and MLVA are DNA length-based typing methods that provide increased resolution, but their reliance on electrophoretic band patterns translates to intrinsically limited data portability. Consequently, the DNA sequence-based methods MLST and cgMLST, analyzing SNPs in 7 loci or the core genome, respectively, have emerged as the methods of choice in recent years. These methods share several limitations, however, relating to cost, technical complexity, and turnaround time that have largely relegated their use to analyzing V. parahaemolyticus evolution and strain dispersal, while their application to outbreak detection and investigation has, to our knowledge, been retrospective only (Ansede-Bermejo et al., 2010; Turner et al., 2013; Haendiges et al., 2015, 2016; Hazen et al., 2015; Xu et al., 2015; Espejo et al., 2017). An additional limitation shared by these DNA-based methods and serotyping is the requirement for a pure culture. The work presented here was driven by the need for a typing method that addresses the issues of cost, complexity, and turnaround time while providing the resolution required for routine strain surveillance and real-time outbreak detection and investigation. PLST is based on conventional PCR amplification and dideoxynucleotide sequence analysis of the one or two genomic loci bioinformatically identified as the most phylogenetically informative. It is technically robust, exemplified by the ability to type directly from crude enrichment cultures. In contrast to whole genome sequences requiring complicated, computationally-intensive analysis, PLST is user-friendly, based on simple clustal alignments and BLASTN searches of publicly available GB databases. Finally, one or two locus-based PLST provides an affordable alternative to other methods since it can be commercially outsourced and hence requires no additional investment in equipment, reagents, or trained personnel.
In the bioinformatic evaluation of PLST candidates, the initial goal was identification of the most polymorphic loci. Both VpMT1 and VpMT2, representing tandem repeat-containing loci on chromosomes 1 and 2, respectively, were exceptional in this regard with North American isolates yielding DI = 0.99. Reassuringly, both VpMT1 and VpMT2 include tandem repeats previously shown to be the most informative components (DI = 0.88–0.96) of MLVA protocols (Kimura et al., 2008; Harth-Chu et al., 2009; Ansede-Bermejo et al., 2010; Jiang et al., 2016); the increased DI associated with PLST reflects sequence-level polymorphism not detected by length-based MLVA. Additional criteria for PLST loci included amenability to amplification and sequencing, stability to laboratory passage, and correlation with available epidemiological and typing data. By these criteria, VpMT1 emerged as the most promising locus for V. parahaemolyticus PLST, with VpMT2 available as needed to provide confirmation and enhanced resolution.
Although the sample number was relatively small, a clear difference was observed between typing directly from oyster enrichments (4 of 13 yielding VpMT1 sequences consistent with V. parahaemolyticus) compared to typing from blue-green colonies on TCBS plates streaked from those enrichments as per FDA laboratory guidelines (1 of 13 yielding V. parahaemolyticus). This likely reflects the ability of PCR to specifically amplify very minor components within complex mixtures and represents an additional advantage (besides the obvious savings in time and effort) of enrichment-based typing. On the other hand, the clinical relevance of V. parahaemolyticus contamination of oysters at very low levels is unclear. In this regard, it should be noted that PLST also provides the capability, via nested PCR, to type directly from unenriched food samples, including oyster liquors (unpublished data). This culture-independent PLST, while less sensitive than enrichment-based PLST, would provide a more realistic measure of contamination levels.
Footnotes
Acknowledgments
We thank Michael A. Watson, USDA-ARS (Dover, DE) for technical assistance. This work was supported by USDA-NIFA SBIR grant no. 12263323 to T.E., and by USDA-ARS intramural funding under CRIS 9072-42000-081-00D to G.P.R. The use of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA. USDA is an equal opportunity provider and employer.
Disclosure Statement
T.E. discloses that he is an employee of MicrobiType LLC, which has commercialized V. parahaemolyticus strain typing services. For G.P.R., no competing financial interests exist.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.