
Abstract
This study evaluated a combined method for the detection of Listeria monocytogenes in mushrooms, involving enrichment and quantitative real-time polymerase chain reaction (qPCR), to improve sensitivity and reduce detection time. The growth of L. monocytogenes was evaluated in Listeria enrichment broth (LEB) with modified carbon and nitrogen sources, increasing sodium concentrations, and added micronutrients. Primers targeting the L. monocytogenes iap (iap1 and iap2), hlyA (hlyA1–hlyA6), and prfA (prfA1–prfA4) genes were developed and their sensitivity and specificity were evaluated. The greatest increase in L. monocytogenes cell count was observed after 6-h incubation at 30°C in LEB+2 × FAC (LEB plus 20 mL/L ferric ammonium citrate), where cell count increased by 1.4 log CFU (colony-forming unit)/mL, compared with 0.9 log CFU/mL in LEB (p < 0.05). iap2 primers targeting the iap gene showed high specificity and were the most sensitive among those tested, with a detection limit of 2 log CFU/mL in LEB medium, 3.1 log CFU/g in golden needle mushroom, and 3.5 log CFU/g in large oyster mushroom. When applied to detection in golden needle mushrooms, a combination of 3-h incubation in LEB+2 × FAC medium and qPCR analysis with iap2 primers permitted detection of L. monocytogenes, even at an inoculum of 1 log CFU/g. Similarly, in large oyster mushrooms, 10-h enrichment in LEB+2 × FAC medium resulted in a cell count of 3.7 log CFU/g. These results indicate that a combined detection method, using LEB+2 × FAC medium for enrichment followed by qPCR with iap2 primer pair, can reduce enrichment time and improve the sensitivity and specificity of L. monocytogenes detection in mushrooms.
Introduction
The Food and Agriculture Organization (FAO) of the United Nations reports that mushroom production reached ∼20 million tons worldwide in 2017 (FAOSTAT, 2019); mushroom consumption is high due to their nutritional value and agreeable flavor. In the United States, the consumption of fresh mushrooms was ∼1.8 kg per capita in 2018, which was higher than consumption (1.1 kg) of garlic (Statista, 2019). However, in 2016, Listeria monocytogenes was detected in golden needle mushrooms exported from Korea to Europe (Lee et al., 2018). Although L. monocytogenes is a known contaminant of meat and processed meat products, such as ham and sausage (Rodrigues et al., 2017), the Interagency Food safety Analytics Collaboration (IFSAC) reported that 75% of all foodborne outbreaks in 2016 are caused by contaminated dairy and fruits (IFSAC, 2018). Consequently, some studies have reported the occurrence of L. monocytogenes in edible mushrooms in several countries (Cordano and Jacquet, 2009; Venturini et al., 2011; Viswanath et al., 2013).
L. monocytogenes is a Gram-positive bacterium that colonizes the human gastrointestinal tract, which can cause systemic listeriosis, with a mortality rate of up to 30% (Vázquez-Boland et al., 2001; Wulff et al., 2006). The primary cause of human listeriosis is intake of contaminated food (Jalali and Abedi, 2008). Standard methods for the detection of L. monocytogenes are culture based, which requires more than 3 days for identification (Ministry of Food and Drug Safety, 2017). With culture-based methods, it is difficult to distinguish different Listeria species and thus, further identification procedures are required. In addition, L. monocytogenes can be present in a viable, but nonculturable state in food, requiring a two-step enrichment procedure. The primary step resuscitates the target bacteria, and the enrichment step suppresses the competitive flora and increases cell counts of the target bacteria such as Campylobacter spp. and Escherichia coli O157:H7 to 104–106 CFU (colony-forming unit)/mL (Jasson et al., 2010; Dwivedi and Jaykus, 2011). To address these limitations, various detection methods have been developed; however, it has been difficult to find the most suitable method for L. monocytogenes detection in each food matrix (Zunabovic et al., 2011). For example, various molecular biological methods, such as polymerase chain reaction (PCR), have been developed; however, the drawbacks of PCR include negative effects of food matrices on amplification (Koo and Jaykus, 2003; Jasson et al., 2010; Yeni et al., 2014). Specifically, Rijpens and Herman (2002) reported that the sensitivity of quantitative real-time polymerase chain reaction (qPCR) applied to a food matrix is low when compared with other enumeration methods. In addition, mushrooms exhibit a specific matrix effect, making bacterial detection more difficult (Kataoka et al., 2012). It is, thus, necessary to develop a rapid and sensitive method to detect L. monocytogenes in mushrooms.
Therefore, the objective of this study was to develop a L. monocytogenes detection method using a modified enrichment medium qPCR.
Materials and Methods
Preparation of Listeria inocula
Five L. monocytogenes strains (BL5-1, D2L3-1, HL1-1, IL1-1, and PL1-1), Listeria innocua G1-1, and Listeria welshimeri BKW11 (provided by the National Institute of Agricultural Sciences) were incubated in tryptic soy broth plus 0.6% yeast extract (TSBYE; Becton, Dickinson and Company, Franklin Lakes, NJ) at 30°C for 24 h. Following incubation, the 100-μL aliquots of the cultures were inoculated into 10 mL TSBYE, and incubated at 30°C for 24 h. Each subculture was centrifuged at 1912 g and 4°C for 15 min, and the resulting cell pellets were washed twice with phosphate-buffered saline (PBS; pH 7.4; KH2PO4 0.2 g/L, Na2HPO4 1.5 g/L, NaCl 8.0 g/L, KCl 0.2 g/L). Subsequently, each cell pellet was resuspended in 10 mL PBS. The cell suspensions of five L. monocytogenes strains were combined to be used as inoculum. Each suspension of L. innocua G1-1 and L. welshimeri BKW11 was also used as inoculum.
Preparation of E. coli, Staphylococcus aureus, and Salmonella Typhimurium inocula
E. coli NCCP14038, Staphylococcus aureus ATCC27664, and Salmonella Typhimurium NCCP11116 were incubated in 10 mL tryptic soy broth (Becton, Dickinson and Company) at 37°C for 24 h and then processed as per Listeria spp.
Preparation of the enrichment media
Media were modified by the addition or replacement of different components to maximize L. monocytogenes enrichment. Listeria enrichment broth (LEB; Becton, Dickinson and Company) was used as a control medium. As shown in Table 1, modified LEB media were prepared by replacing or adding carbon source, nitrogen source, sodium, and micronutrients in LEB, for which the standard recipe was for 1 L. The standard carbon source (glucose) was replaced by sucrose, galactose, or sucrose+galactose. Modified nitrogen sources were as follows: yeast extract, beef extract, or yeast extract+beef. Sodium chloride concentrations were by increased amounts. Also, some micronutrient sources were evaluated (KH2PO4, KCl, NaHCO3, Na2PO4, CaCl, and ferric ammonium citrate [FAC]). All media were left at 25°C ± 2°C after sterilization, and they were used when the media temperature became 25°C ± 2°C. Prepared L. monocytogenes inoculum was inoculated into each enrichment medium at 2 log CFU/mL, and incubated at 30°C for 6 h. The cultures were analyzed at 0, 3, and 6 h. At each time point, cultures were serially diluted with buffered peptone water (BPW; Becton, Dickinson and Company), and spread on tryptic soy agar plus 0.6% yeast extract (TSAYE; Becton Dickinson and Company). TSAYE plates were incubated at 30°C for 48 h, and then colonies were manually counted.
Information of Added Ingredients and Concentration for Modifying Enrichment Medium
FAC, ferric ammonium citrate; LEB, Listeria enrichment broth.
Primer design
To detect L. monocytogenes in the enriched samples, the full sequences of the hemolysin gene (hlyA), p60 gene (iap), and regulatory gene (prfA) genes were obtained from the National Center for Biotechnology Information (NCBI). Primers were designed to hlyA, iap, and prfA using OligoPerfect™ Designer, an online-based software provided by Thermo Fisher Scientific (MA;
Primer specificity
Aliquots (1 mL) of L. monocytogenes strain mixture, L. innocua G1-1, L. welshimeri BKW11, E. coli NCCP 14038, S. aureus ATCC 27664, and Salmonella Typhimurium NCCP 11116 inocula were centrifuged at 8500 g and 4°C for 10 min, and the supernatants were discarded. DNA was extracted from the pellets using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Extracted DNA was then used to evaluate the specificity of each primer by qPCR in a Rotor-Gene Q instrument (Qiagen), using the Rotor-Gene SYBR® Green PCR Kit (Qiagen). A 25 μL reaction mixture was used, comprising 2 μL template DNA, 12.5 μL 2 × Rotor-Gene SYBR Green PCR master mix, 5.5 μL RNase-free water, 2.5 μL forward primer, and 2.5 μL reverse primer. The amplification conditions were: 5 min preheating at 95°C, followed by 35 cycles of 5 s denaturation at 95°C, 10 s annealing at 56°C, and 10 s elongation at 72°C. Assay specificity was estimated based on the melting characteristics of the amplified products (Nolan et al., 2013). L. monocytogenes IL1-1 DNA was used as a melting temperature (Tm) positive control; the Tm of other samples were then compared with this, to determine positivity.
Primer sensitivity
The primer pair demonstrating the best specificity was selected and analyzed to evaluate sensitivity. Serial dilutions of L. monocytogenes IL1-1 were performed in LEB from 5 log CFU/mL to <1 log CFU/mL and DNA was extracted from each dilution as described before. In addition, 100 μL of the IL1-1 dilutions were inoculated into 20 g golden needle mushrooms and large oyster mushrooms in sterile sample bags (3M™, St. Paul, MN). Inoculated mushroom samples were left for 10 min under a laminar flow cabinet for the cell attachment and then 40 mL BPW was added. Samples were then macerated using a BagMixer instrument (Interscience, St. Nom, France) for 1 min, and DNA was extracted (as before) from 3-mL aliquots of the homogenates and amplified by qPCR. Cycle threshold (CT) values, determined by Rotor-Gene Q series software (Stratagene, La Jolla, CA), were used to evaluate primer sensitivity.
Application of the detection methods in mushroom samples
Large oyster mushrooms and golden needle mushrooms (20 g) were placed in sterile bags, and inoculated with 100 μL of L. monocytogenes inoculum to obtain ∼1 log CFU/g mushroom. Samples were left for 10 min under a laminar flow cabinet, to allow cell attachment and then 40 mL LEB medium or modified LEB+2 × FAC (20 mL FAC per liter) medium were added into each sample bag. Samples were then macerated as before and homogenates were incubated at 30°C for 0, 3, 6, 9, 10, 11, and 12 h for enrichment. DNA was extracted from 3 mL of each enriched culture and amplified as before. Enriched samples were also serially diluted with BPW, and dilutions were spread on PALCAM agar (Oxoid, Basingstoke, United Kingdom). Plates were then incubated at 30°C for 48 h, after which L. monocytogenes was enumerated in each enrichment medium and incubation time. To evaluate if the developed detection method can be applied to mushrooms, five golden needle mushrooms and five large oyster mushrooms were purchased from a market and analyzed with the developed method. In addition to validate the result, 25-g portions of mushroom samples were placed to 225 mL of LEB medium in sterile sample bags and pummeled by a pummeler (BagMixer®, Saint Nom, France) for 1 min. The mixtures were then enriched at 30°C for 48 h, and the enriched aliquots were streaked on the PALCAM agar. After the incubation at 30°C for 48 h, gray/green colonies with a black halo on PALCAM agar further analyzed for 16s rRNA sequencing (Suau et al., 1999) to determine if the colonies were positive.
Statistical analysis
L. monocytogenes cell counts were analyzed using a general linear model procedure with SAS® version 9.3 (SAS Institute, Inc., Cary, NC). The least square means were used for mean comparison among enrichment broths and among enrichment time by pairwise t-test at α = 0.05.
Results and Discussion
Modified enrichment broth
Neither addition of carbon or nitrogen sources, nor increasing salt concentration in LEB medium resulted in increased L. monocytogenes cell counts, when compared with LEB medium (data not shown). Cell counts in standard LEB medium increased by 0.6 log CFU/mL after 6-h incubation at 30°C (p < 0.05), and by 1 log CFU/mL in LEB+FAC (10 mL of FAC per liter) medium (p < 0.05; Table 2). This suggests that the use of FAC in LEB medium improved the efficiency of L. monocytogenes enrichment and therefore different concentrations were evaluated (10 mL/L [FAC], 20 mL/L [2 × FAC], 30 mL/L [3 × FAC], and 40 mL/L [4 × FAC]). The greatest enrichment effect was seen with LEB+2 × FAC medium, which resulted in a cell count of 3.2 log CFU/mL after 6 h, representing an increase of 1.4 log CFU/mL (p < 0.05; Table 3). There was no difference in cell counts between LEB+2 × FAC medium and LEB+4 × FAC medium after 3- and 6-h of incubation. Hence, LEB+2 × FAC medium was selected as the most efficient enrichment medium. According to McLaughlin et al. (2011), L. monocytogenes can acquire and utilize iron to support its growth and survival in many diverse environments. Therefore, high concentration of iron in FAC may enrich L. monocytogenes growth more than standard LEB. This may reduce the time needed to enrich L. monocytogenes to the concentrations required for detection by qPCR. Combining enrichment in LEB+2 × FAC medium with qPCR may therefore be effective in minimizing the length of the enrichment step when detecting L. monocytogenes in mushrooms.
Change in Listeria monocytogenes Cell Counts (Log CFU/mL) Following Enrichment in Each of the Enrichment Broth at 30°C
Indicates significant difference between the same column (p < 0.05).
CFU, colony-forming unit; FAC, ferric ammonium citrate; LEB, Listeria enrichment broth.
Listeria monocytogenes Cell Counts (Log CFU/mL; Mean ± Standard Deviation) Following Enrichment in Different Media at 30°C at Different Time Points
2 × FAC: 20 g of ferric ammonium citrate per liter.
3 × FAC: 30 g of ferric ammonium citrate per liter.
4 × FAC: 40 g of ferric ammonium citrate per liter.
Means with the same column with different superscript capital letters and same row with different superscript small letters are significantly different (p < 0.05).
CFU, colony-forming unit; FAC, ferric ammonium citrate; LEB, Listeria enrichment broth.
Selection of effective primers
A total of 12 primers were designed, 6 were targeted to the hlyA gene (hlyA1–hlyA6), 4 to the prfA gene (prfA1–prfA4), and 2 to the iap gene (iap1 and iap2) of L. monocytogenes (Table 4). For L. monocytogenes IL1-1, the Tm of the hlyA2 and iap2 primers were 81°C and 83°C, respectively, and these Tm values were used as a positive control. In addition, the DNA isolated from L. innocua, L. welshimeri, S. aureus, Salmonella Typhimurium, and E. coli were not amplified with the primers. This result showed that the hlyA2 and iap2 primers were specific for L. monocytogenes. However, the remaining 10 primer pairs were not specific for L. monocytogenes, resulting in the same Tm for all three Listeria species tested. Both hlyA2 and iap2 primer pairs demonstrated the same detection limit (2 log CFU/mL) in TSBYE medium (Table 5). When tested with DNA extracted from large oyster mushrooms inoculated with L. monocytogenes, iap2 primers were more sensitive than hlyA2 primers, where the limits of detection were 3.5 log CFU/g and 5.4 log CFU/g, respectively (Table 6). In addition, the detection limit of iap2 primers in the golden needle mushrooms were 3.1 log CFU/g and that of hlyA2 primers were 4.6 log CFU/g. This result indicates that qPCR amplification of DNA using iap2 primers can be used for the specific and sensitive detection of L. monocytogenes in mushrooms. Therefore, cultivation in LEB+2 × FAC enrichment broth, followed by amplification with iap2 primers was evaluated for the detection of L. monocytogenes in mushrooms.
Primers Used in This Study for Detecting Listeria monocytogenes
F: forward sequence (5′–3′) of the primers; R: reverse sequence (5′–3′) of the primers.
Tm, melting temperature in qPCR.
qPCR, quantitative real-time polymerase chain reaction.
Primer Sensitivity for Detection of Listeria monocytogenes in Broth Culture
CFU, colony-forming unit; NA, not amplified with the sample; SD, standard deviation.
Primer Sensitivity for Detection of Listeria monocytogenes in Mushrooms
CFU, colony-forming unit; NA, not amplified with the sample; SD, standard deviation.
Application of the selected detection method in mushroom samples
Immediately after inoculation in golden needle mushrooms, L. monocytogenes was detected by qPCR in two out of four golden needle mushroom samples (Table 7). After 3-h enrichment, L. monocytogenes was detected in all four samples, with an average CT value of 29.05 (Table 7). Furthermore, at this time point, the cell count was 1.6 log CFU/g in LEB+2 × FAC, which was higher (p < 0.05) than in LEB medium (Table 7). There was also a significant difference in the cell counts between LEB and LEB+2 × FAC media after 6-h enrichment, where cell counts increased by 0.7 log CFU/g and 1.1 log CFU/g, respectively (p < 0.05) (Table 7). L. monocytogenes was detected in all four samples and the mean CT value was 30.36 after 6-h enrichment (Table 7). The specificity and sensitivity of hlyA2 and iap2 primers for detection of L. monocytogenes in golden needle mushroom was evaluated by qPCR. The positive control (L. monocytogenes IL1-1) is represented in Figure 1 indicated by an arrow in each case that showed highest parabolic graph. DNA extracted from enriched mushroom samples and amplified with iap2 primers showed the same peak as the control (Fig. 1a), indicating the same Tm; however, use of hlyA2 primers resulted in a different peak, indicating incorrect amplification (Fig. 1b). Although hlyA primers have been used to detect L. monocytogenes in broth cultures previously (Burall et al., 2011), this result indicates that iap2 primers are more suitable than hlyA2 primers for the detection of L. monocytogenes in mushrooms. Klein and Juneja (1997) also reported that targeting the iap gene-specific mRNA yielded better detection limits with qPCR, compared with mRNA from the hlyA and prfA genes. Furthermore, Liu et al. (2015) reported that the iap gene offered greater specificity than the 16S rRNA gene for the detection of L. monocytogenes, because of the high similarity between L. monocytogenes and L. innocua 16S rRNA sequences (99.07%). Taken together, these data suggest that appropriate conditions for the detection of L. monocytogenes in golden needle mushroom include a 3-h incubation in LEB+2 × FAC medium followed by qPCR analysis with iap2 primers, even for concentrations at 1 log CFU/g.
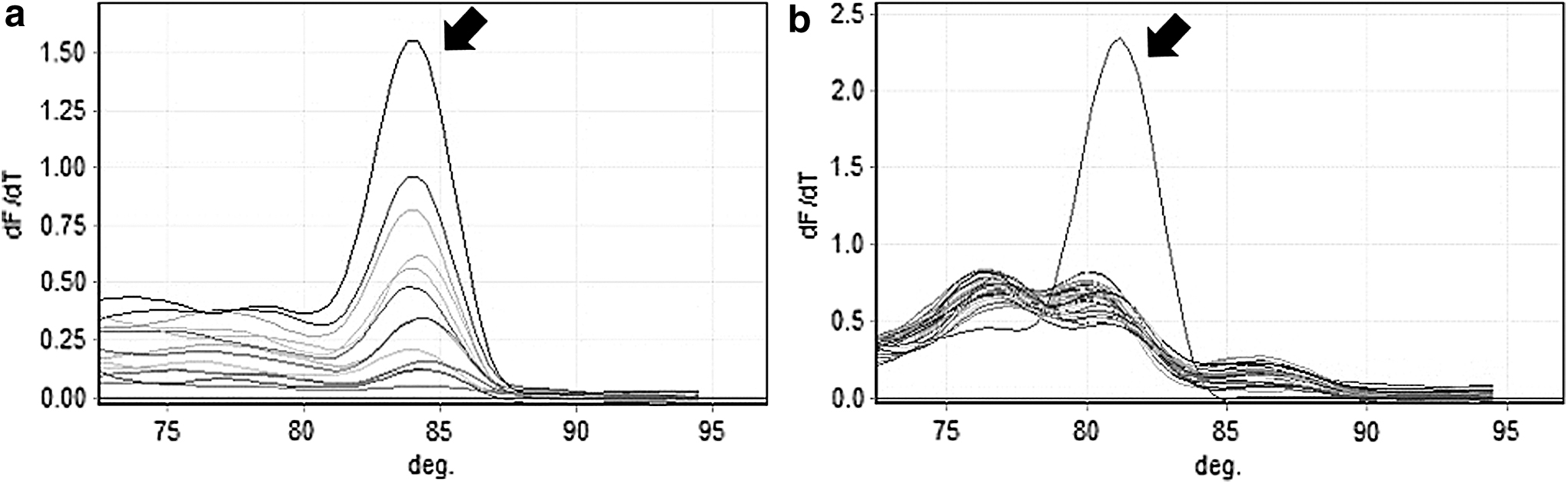
Melting curves following qPCR amplification of Listeria monocytogenes DNA from golden needle mushrooms with iap2 primers
Listeria monocytogenes Cell Counts (Log CFU/g; Mean ± Standard Deviation) and Quantitative Real-Time Polymerase Chain Reaction Results (Cycle Threshold and the Number of Positive Samples) in Golden Needle Mushroom Samples Enriched in Listeria Enrichment Broth and Listeria Enrichment Broth+2 × Ferric Ammonium Citrate
Means with the same column with different superscript capital letters and same row with different superscript small letters are significantly different (p < 0.05).
Positive sample as determined by a melting temperature (Tm), matching that of the positive control.
CFU, colony-forming unit; FAC, ferric ammonium citrate; LEB, Listeria enrichment broth; NA: not amplified with the sample; qPCR, quantitative real-time polymerase chain reaction.
In large oyster mushroom, 10-h enrichment in LEB+2 × FAC medium led to a significantly higher L. monocytogenes cell count (3.7 log CFU/g) than those enriched in LEB medium (3.1 log CFU/g; p < 0.05; Table 8). This suggests that the optimal conditions for the detection of L. monocytogenes in large oyster mushroom at 1 log CFU/g include enrichment with LEB+2 × FAC for 10 h at 30°C, followed by qPCR using iap2 primers. Rossmanith et al. (2006) reported that 24-h enrichment, followed by qPCR targeting the prfA gene, provided detection limits of 7.5 CFU/25 mL in raw milk and 9, 1, 1 CFU/15 g in salmon, pate, and cheese contaminated with L. monocytogenes, respectively. Our method requires less time and is effective at a similar level of contamination. We found that longer enrichment times were required for positive results in large oyster mushrooms than in golden needle mushrooms. According to the study of Ahn et al. (2006), the total content of polyphenol was much higher in the large oyster mushroom than that of the golden needle mushroom. Also, polyphenols have antimicrobial activities against Gram-positive bacteria (Papuc et al., 2017). Therefore, polyphenols in large oyster mushroom may retard L. monocytogenes enrichment.
Listeria monocytogenes Cell Counts (Log CFU/g; Mean ± Standard Deviation) and Quantitative Real-Time Polymerase Chain Reaction Results (Cycle Threshold and the Number of Positive Samples) in Large Oyster Mushroom Samples Enriched in Listeria Enrichment Broth and Listeria Enrichment Broth+2 × Ferric Ammonium Citrate
Means with the same column with different superscript capital letters and same row with different superscript small letters are significantly different (p < 0.05).
Positive sample as determined by a melting temperature matching that of the positive control.
CFU, colony-forming unit; FAC, ferric ammonium citrate; LEB, Listeria enrichment broth; NA, not amplified with the sample; qPCR, quantitative real-time polymerase chain reaction.
Applied in real mushrooms, all the large oyster mushroom samples were negative for L. monocytogenes in qPCR and culture methods. Among the golden needle mushrooms, L. monocytogenes was positive in one sample. The culture method detected similar colonies in three different samples, and up to 16s rRNA analysis showed that they were not all L. monocytogenes. Therefore, the method of this study was found to be applicable to real mushroom samples.
Conclusion
LEB+2 × FAC medium can reduce the enrichment time, compared with LEB. The designed primers (iap2 primers), targeting iap gene has higher sensitivity and specificity for L. monocytogenes than other primers. Therefore, a combined method using LEB+2 × FAC medium for enrichment and qPCR with iap2 primers can reduce the time to detect L. monocytogenes in mushrooms with improved sensitivity and specificity.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by a grant (PJ01353605) from the Rural Development Administration, Republic of Korea.