
Abstract
Shiga toxin-producing Escherichia coli (STEC) consists of a group of diverse strains differing greatly in genetic make-up and pathogenicity potential. Here, we investigated production of Shiga toxins (Stxs) in a bovine isolate carrying multiple Shiga toxin genes (stxs) after exposure to several antibiotics commonly used in food animals. Strain RM10809-C3 was co-isolated with a STEC O145:H28 strain from cattle feces near a leafy greens-growing region in California. The genome of RM10809-C3 is composed of a 5,128,479-bp chromosome and a 122,641-bp plasmid, encoding 5108 coding sequences. Strain RM10809-C3 belongs to serotype O22:H8 and is clustered together with two STEC O168:H8 food isolates using either multilocus sequence type or core genome-based phylogenetic analysis. Six intact prophages were identified in the genome of RM10809-C3, among which prophage 4 contained two sets of stx 2d; whereas prophage 9 carried one set of stx 1a. Increased production of Stx1 was detected in RM10809-C3 after exposure to mitomycin C and enrofloxacin, but not in cells exposed to tetracycline. In contrast, Stx2 remained undetectable in cells treated with any of the antibiotics examined. Comparison of Stx-converting prophages in strain RM10809-C3 with those in strain EDL933 revealed altered stx2 promoters in RM10809-C3, including deletion of the late promoter P R′ and the mutations in qut, the binding site of antitermination protein Q. In contrast, both P R′ and qut within the promoter of stx1 in RM10809-C3 were identical to the corresponding one in EDL933. Further, the protein Q encoded by Stx1-prophage in RM10809-C3 exhibited >94% identity with either of the two EDL933 protein Q; whereas both protein Q encoded by Stx2-prophage in RM10809-C3 were distantly related to any of the EDL933 protein Q. Natural silence of Stx2 production in strain RM10809-C3 emphasizes that not only the stx coding regions but also their regulatory factors are important in STEC risk assessment.
Introduction
Shiga toxin-producing Escherichia coli (STEC) consists of a group of genetically and phenotypically diverse E. coli strains that differ greatly in pathogenicity. Some STEC strains can cause severe diseases, whereas others are only associated with mild diarrhea or no disease at all (Coombes et al., 2011). Such variation is attributed in part to the difference in their genetic makeup. To date, studies have been centered on several major serotypes such as O157:H7 and the “big six” as they are frequently associated with human diseases (Scallan et al., 2011). These clinically important STEC strains are believed to have evolved from cattle-associated STEC strains that were not highly pathogenic to humans (Koudelka et al., 2018).
Shiga toxin (Stx), a major virulence factor in STEC, belongs to a group of bacterial AB5 protein toxins that inhibit protein synthesis in eukaryotic cells (Endo and Tsurugi, 1987; Nataro and Kaper, 1998; Melton-Celsa, 2014). Stx1 and Stx2 are the two major antigenic types, both of which contain several subtypes. STEC strains that produce Stx1a, 2a, 2c, and/or 2d are often associated with severe diseases (National Advisory Committee on Microbiological Criteria for Foods, 2019). The stx genes are organized as an operon, stxAB, located in the late gene region of a temperate lambdoid phage, between the antitermination protein Q gene and the lysis gene cassettes; therefore, the expression of stx is likely under control of the phage regulatory scheme (Schmidt, 2001; Waldor and Friedman, 2005). Under lysogenic state, stx remains silent; however, when the phage lytic program is activated, the CI repressor is inactivated and the transcription of stx is initiated at late promoter P R′ when the antitermination protein Q binds to the Q protein utilization site (qut). The regulator cascade of stx is similar to that of bacterial SOS response; thus, agents that produce single-stranded DNA in STEC cells would lead to induction of Stx-prophage and, consequently, increase Stx production (Kimmitt et al., 2000). Recent research revealed that stresses encountered by STEC in the human intestine and in food production environments could induce Stx-prophages (Wagner et al., 2001; Aertsen et al., 2005; Fang et al., 2017). Induction of Stx-prophage leads to lysis of E. coli cells and release of Stx-phages, which can transfer stx to other bacteria via transduction, promoting emergence of new STEC strains (Acheson et al., 1998; Zhang et al., 2000).
Cattle are the main reservoir for STEC. Addition of sub-therapeutic antibiotics to the feed of food-producing animals for growth promotion and disease prevention has become a common agricultural practice in the United States. Such practice may not only affect antibiotic resistance but also facilitate the dissemination of virulence factors such as Stx via bacteriophages (Tamang et al., 2017). Increased production of Stx-phages as well as Stx was detected in cells exposed to sub-inhibitory concentrations of olaquindox and carbadox (Kohler et al., 2000). Transduction of E. coli cattle isolates by Stx-phage was reported when STEC O157:H7 cells were induced by oxytetracyline or chlortetracycline (Kim et al., 2016). Stx-phages have been detected not only in cattle gastrointestinal tracts but also in the water polluted with feces, food samples, and even stool samples from healthy individuals (Martinez-Castillo and Muniesa, 2014), implying that stx genes are widespread in the natural environment.
Strain RM10809-C3 was co-isolated from bovine feces with an STEC O145:H28 strain described in our previous study (Carter et al., 2016). It carries stx genes encoding Stx1a and the mucus-activatable Stx2d, an Stx2 subtype known to cause hemolytic uremic syndrome (HUS) in infected humans (Bielaszewska et al., 2006). In this study, we examined the genomic signatures of RM10809-C3 and of Stx-prophages by comparative genomics analyses. We further determined whether the antibiotics that are commonly used in livestock could increase the Stx production in RM10809-C3.
Materials and Methods
Bacterial strains
Strain RM10809-C3 was isolated from cattle feces as previously described (Cooley et al., 2013). This strain is motile and produces curli fimbriae at 37°C, but not at 26°C.
Genome sequencing and annotation
Bacterial DNA was extracted from the exponential phase culture grown in LB broth. Cells were lysed with SDS followed by sequential treatment with RNase A and proteinase K. The DNA was first precipitated in a sodium acetate/ethanol solution, and then purified by phenol/chloroform extraction, followed by the final ethanol precipitation. The purified DNA was re-suspended in Qiagen Buffer EB (QIAGEN). The SMRTbell™ library was prepared following the protocols in “Procedure & Checklist-20 kb Template Preparation Using BluePippin™ Size-Selection System” (Pacific Biosciences) and run on a PacBio RS II with on-plate concentration at 0.3 nM, P6/C4 sequencing chemistry, MagBead One Cell Per Well v1 collection protocol, and 360-min data collection mode. Sequence reads were assembled with Hierarchical Genome Assembly (HGAP) version 3.0, and the genome was annotated by using the NCBI Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP). The complete genome was deposited in GenBank with accession numbers CP023164 and CP023165.
Comparative genomic analyses
Phylogeny of RM10809-C3 was determined by core genome in EDGAR (Blom et al., 2009, 2016) or by in silico multilocus sequence type (MLST) (Wirth et al., 2006). A total of 80 genomes representing 54 serotypes of STEC were retrieved from GenBank (Supplementary Table S1). The MLST loci (adk, fumC, gyrB, icd, mdh, purA, and recA) were extracted from each genome. The concatenated sequences were aligned by using ClustalW, and the phylogenetic tree was generated by using PhyML with default setting in Geneious Prime®. Serotyping was determined by BLAST search of GenBank and confirmed with SerotypeFinder 2.0 (Joensen et al., 2015). Virulence genes were determined by using the VirulenceFinder 2.0 (Joensen et al., 2014). Stx-prophages were identified by using PHASTER (Arndt et al., 2016). A neighbor-joining tree of Stx-prophages was generated in Geneious Prime by using Geneious alignment with the following parameter: genetic distance model, Jukes-Cantor; Resampling method, bootstrap; and number of replicates, 10,000.
Induction of Stx
Stx production was examined after exposure of cells to a sub-inhibitory concentration of enrofloxacin or tetracycline. Mitomycin C was included as a control. The minimal inhibition concentration (MIC) of each antibiotic was determined by using the established guidelines for the broth micro-dilution method (Wiegand et al., 2008), except that LB broth was used. The culture in exponential-growth phase (OD600 ∼0.4–0.5) was divided into two 5-mL subcultures: One was added with the antibiotic, and the control was added to the same volume of water or the solvent used for preparation of the antibiotic. Cultures were further incubated overnight at 37°C for 24 h, followed by centrifugation at 7200 × g for 10 min to remove cells. The supernatants were filtered through low-protein-binding 0.22-μm-pore-size membrane filters (Millex-GP PES; Millipore) and stored at −20°C for Stx quantification.
Quantification of Stx
Monoclonal antibody (MAb) pairs were used to establish serotype-specific sandwich ELISA for Stx1a and Stx2a, and standard curves were used to obtain toxin concentration from unknown samples as previously described with some modifications (He et al., 2016). The Stx2a-antibody was shown to be reactive to the Stx2d. Briefly, capture MAbs were immobilized on black high-binding 96-well plates (Nunc) at 1 μg/mL in 0.1M carbonate buffer (pH 9.4), washed in Tris-buffered saline with 0.1% Tween-20 (TBST pH 7.2), and blocked in TBST containing 0.1% IgG-free bovine serum albumin (BSA; Jackson ImmunoResearch, PA). The paired detector MAbs for each assay was conjugated to horseradish peroxidase at a 1:2 stoichiometry (Lightning-link; Innova Biosciences, United Kingdom), used at 1 μg/mL with enhanced chemiluminescence (Neogen, MI) detection of toxins reported in counts per second by using a Victor X3 luminometer (PerkinElmer, MA). All reactions were performed at room temperature with a minimum of three data replicates. Purified Stx1a and Stx2a proteins diluted in TBST-BSA were used to generate standard curves, and no cross-reactivity between the two toxin serotypes was observed. Standard curves were analyzed by four parameters logistic with dynamic curve fitting, and the concentration of toxins in unknown samples was determined from the corresponding standard curve (Stx1a EC50 = 4.8 ng/mL; Hillslope = 1.03; Stx2a EC50 = 13.0 ng/mL; Hillslope = 1.8). The difference in Stx production between the control and the corresponding antibiotic-treated sample was assessed by unpaired t-test with 95% confidence interval by using Prism 7.0 (GraphPad Software).
Results
Genomic signature of RM10809-C3
The RM10809-C3 genome is composed of a 5,128,479 bp chromosome and a 122,641 bp plasmid, with the average GC content of 50.9% and 49.4%, respectively. It encodes 5108 coding sequence (CDS), 22 rRNA, and 93 tRNA (Table 1). RM10809-C3 belongs to serotype O22:H8 and has a sequence type 145 and 446 using Michigan and Warwick scheme, respectively. In silico phylo-typing placed this strain in phylo-group B1, a group containing diverse STEC strains (Supplementary Table S1).
Genomic Characteristics of Strain RM10809-C3 in Comparison with Representative Shiga Toxin-Producing Escherichia coli Strains Carrying stx1 and stx2 Genes
Prophages were identified by using PHASTER (
There are six intact and four possible prophage regions in the RM10809-C3 chromosome (Fig. 1A). Prophage 9 is 67,657 bp in length, containing stx 1a. Prophage 4 is 119,680 bp in length, containing two copies of stx 2d (Fig. 1B). Although both copies of stx 2d are located immediately downstream of tRNA-Met/tRNA-Arg/tRNA-Arg, and share nearly identical CDSs, the upstream sequences including the gene encoding antitermination protein differ greatly (Fig. 1C).
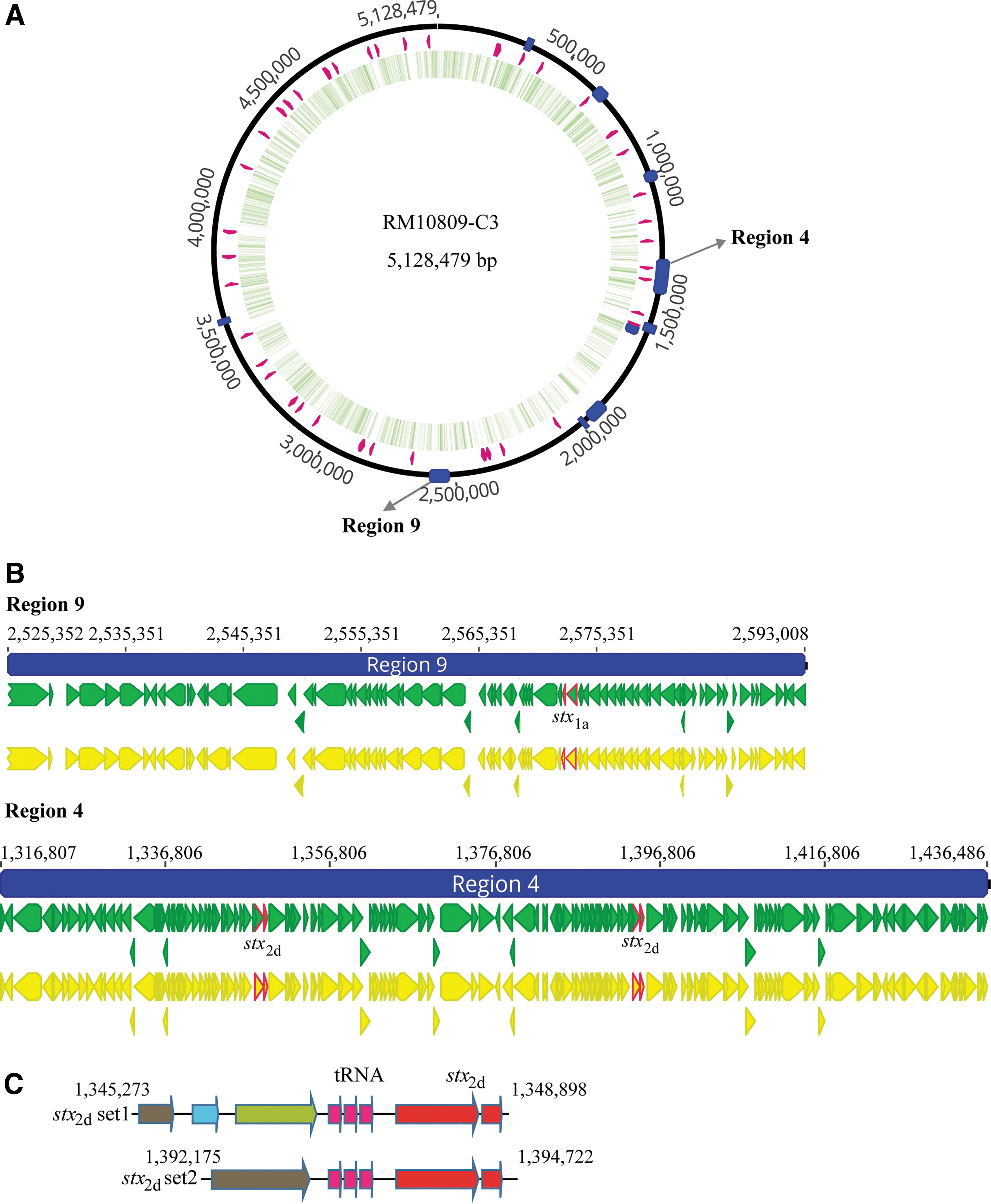
Stx-prophages in RM10809-C3.
In addition to stx, RM10809-C3 carries other known virulence genes, including these encoding functions involved in acid resistance (gad), adherence (iha), attachment (lpfA), and biofilm formation (csgBADEFG), as well as host survival (iss) (Table 2). The large plasmid pRM10809-C3 carries genes encoding enterohemolysin, the extracellular serine protease, and an Iha adherence protein.
Comparative Analysis of Genes Encoding Virulence Factors in Strain RM10809-C3
Virulence genes were determined by using the VirulenceFinder 2.0 (Joensen et al., 2014).
The incompatibility of each plasmid was determined by using the PlasmidFinder-2.0 (Carattoli et al., 2014).
LEE, Locus of Enterocyte Effacement.
Unlike EHEC strains, RM10809-C3 does not possess the Locus of Enterocyte Effacement (LEE) pathogenicity island although it contains several LEE integration sites such as selC tRNA and pheV tRNA (Bertin et al., 2004). However, RM10809-C3 carries a complete Locus of Adhesion and Autoaggregation (LAA) pathogenicity island that was recently identified in an array of emerging STEC strains associated with human diseases (Montero et al., 2017). The LAA is 92,457 bp in length, located on chromosome (3,504,100–3,596,756), and encodes 99 CDS including adhesins and autotransporter adhesion Ag43, transcriptional regulators, as well as type II and type VI secretion proteins, and several transposases.
Phylogeny of strain RM10809-C3
The MLST-based phylogenetic tree grouped RM10809-C3 together with the two O168:H8 strains (Warwick, ST718): One was isolated from lettuce and the other from ground beef (Supplementary Table S1). Further, RM10809-C3 formed a clade with STEC O130:H11 strain 07-4299 and appeared to have evolved from a common ancestor with strains of several serotypes known to cause HUS in humans, including O104:H4, O103:H2, O45:H2, and O113:H21 (Fig. 2). This phylogeny was consistent with that based on a core of 2063 CDS constructed in EDGAR (Data not shown).
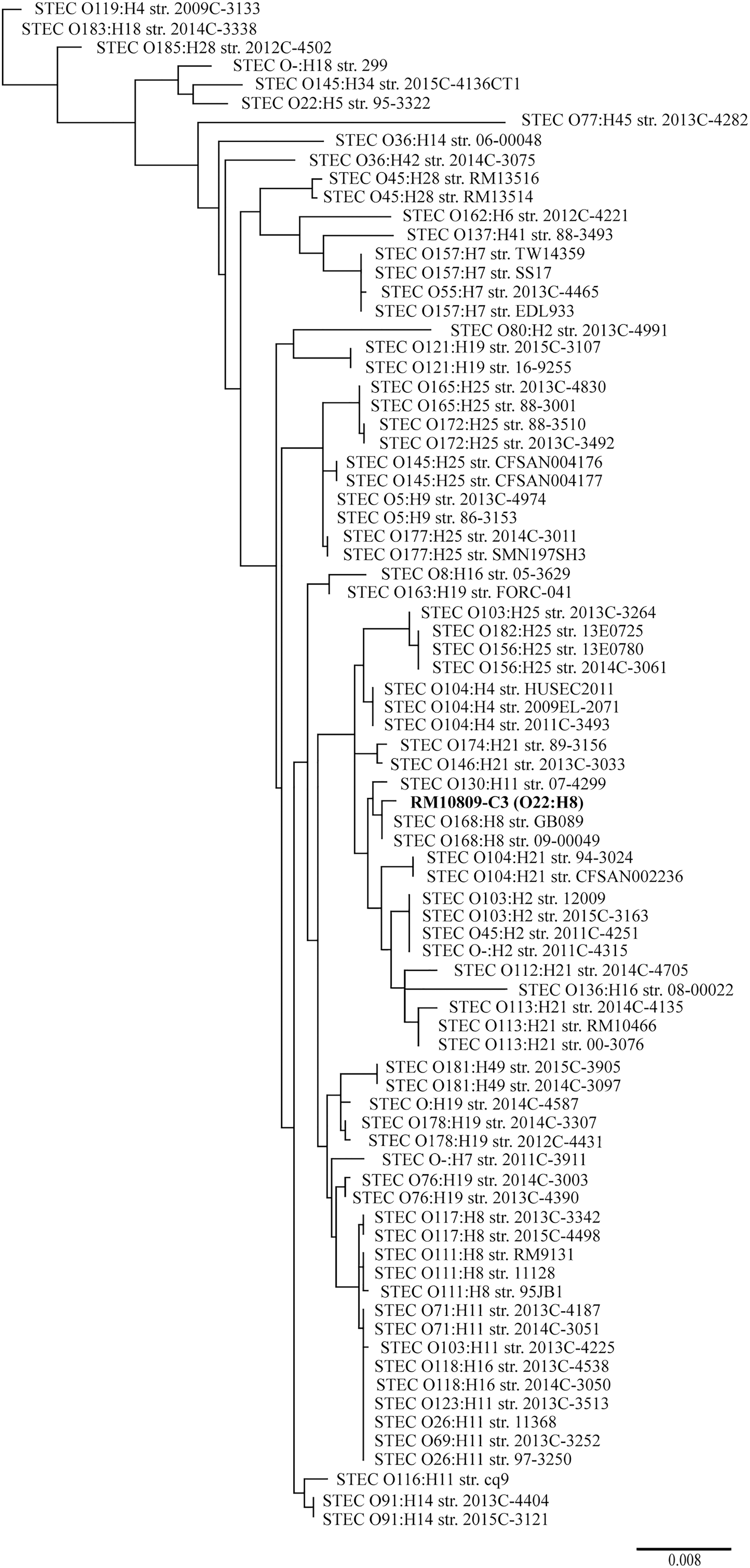
Phylogeny of RM10809-C3. A total of 80 STEC genomes were retrieved from GenBank. The seven MLST loci used in Warwick scheme were extracted from each genome, including RM10800-C3 (In bold). The concatenated sequences were aligned by using ClustalW, and the consensus sequence was used to construct phylogeny tree by using PhyML method in Geneious Prime® as detailed in the Materials and Methods section. Each genome was labeled with serotype followed by the strain name. The GenBank accession number for each genome as well as phylogeny group, ST, and stx subtypes determined in silico were listed in Supplementary Table S1. MLST, multilocus sequence type; ST, sequence type; STEC, Shiga toxin-producing Escherichia coli.
BLAST sequence analysis of the RM10809-C3 plasmid revealed that pRM10809-C3 exhibited the highest similarity with the pEHEC of the two clinical strains 2012C-4431 (O178:H19) and 2014C-4587 (O-:H19), and both were placed in a different clade from that of RM10809-C3 (Fig. 2). The pRM10809-C3 belongs to incompatibility group IncFIB, similar to the pEHEC of strains EDL933 12009, 2013C-4830, and 2012C-4481 (Table 2). The pRM10809-C3 shares five locally collinear blocks (LCBs) with the two pEHEC plasmids (Fig. 3), among which LCB1 contains genes encoding enterohemolysin and exhibits 91.1% and 98.6% identity with that of p2012C-4431 and p2014C-4587. Other conserved functions include extracellular serine protease (LCB2), transposases (LCB3), and conjugative transfer (LCB4 and LCB5).
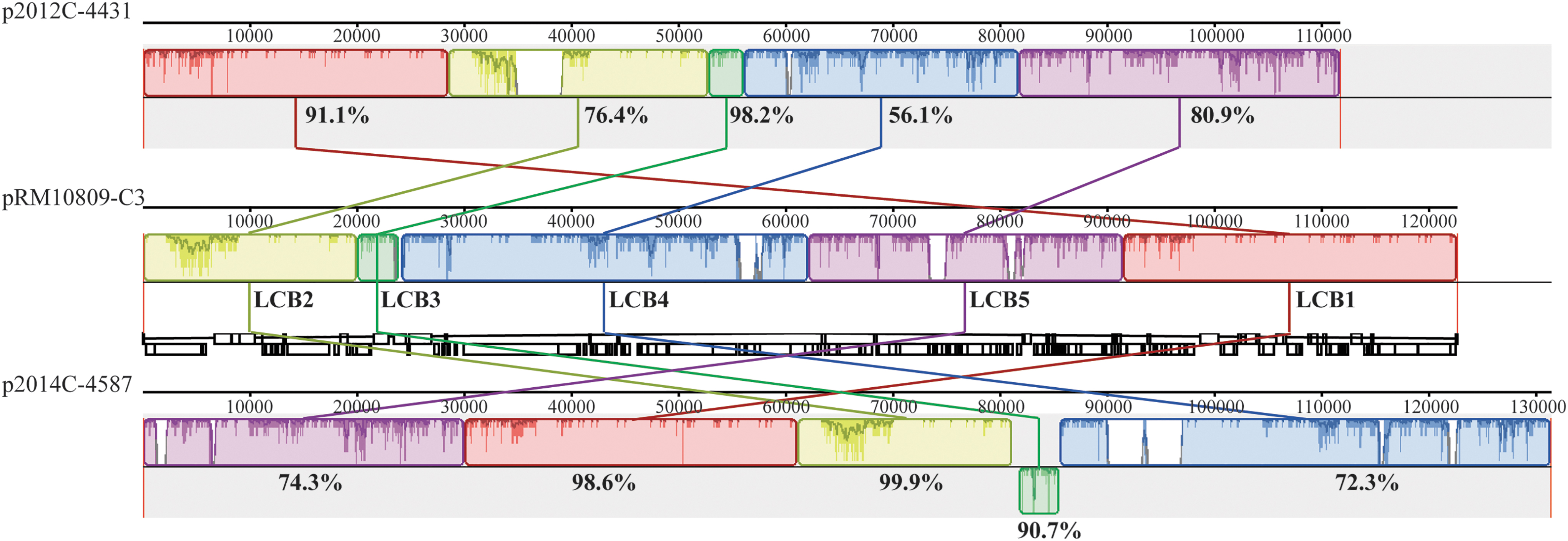
Comparative analysis of RM10809-C3 plasmid pRM10809-C3. The sequence of large plasmid pRM10809-C3 was aligned with the plasmids p2012C-4431 and p2014C-4587 belonging to the STEC strains 2012C-4431 and 2014C-4587, respectively, using progressive Mauve algorithm in Geneious Prime. The numbers indicate the length of DNA segments in bp; the percentage indicates the identity in nucleotide sequences between the two LCBs as detailed in the Materials and Methods section. Each color represents a unique LCB. LCB, locally collinear block; STEC, Shiga toxin-producing E. coli.
Stx-prophages
Cluster analysis revealed that the two Stx-prophages in RM10809-C3 were distantly related (Supplementary Fig. S1). The Stx1-prophage (prophage 9) was closely related to the Stx2-prophages of STEC O104:H21 clinical strains 94-3024 and CFSAN002236; whereas the Stx2-prophage (prophage 4) exhibited the highest similarity with the Stx2-prophage of strain 95-3322 (O22:H5), and it was grouped together with the Stx2-prophages of strains RM10466 (O113:H21) and 2012C-4502 (O185:H28) and the Stx1-prophage of strain 00-3076 (O113:H21).
The Stx1-prophage genome shared high sequence similarity with Stx-prophages in STEC O104:H21 strains 94-3024 and CFSAN002236 (Fig. 4A). Sequence alignment revealed a 3090 bp DNA segment that was unique in RM10809-C3 Stx1-prophage, encoding an Eae protein, an Ead/Ea22-like family protein, an XRE family transcriptional regulator, and a YfdR protein (5′-deoxynucleotidase).
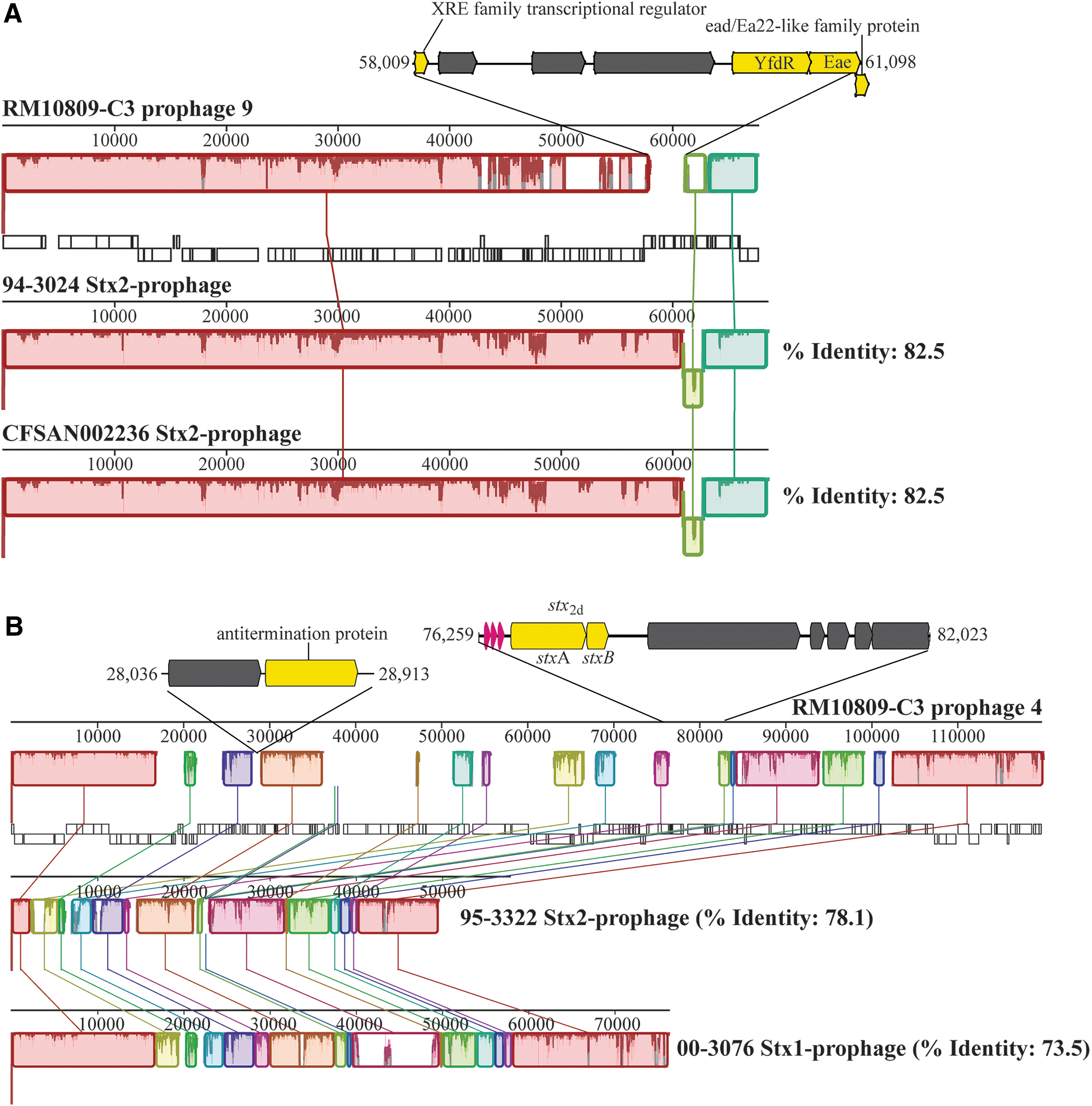
Comparative analysis of Stx-prophages in RM10809-C3.
The Stx2-prophage exhibited a vastly mosaic structure when compared with the closely related prophages (Fig. 4B). A total of 18 LCBs were identified, ranging from 376 to 19,007 bp. The RM10809-C3-specific sequences in Stx2-prophage were incorporated between the LCBs, including a 878-bp DNA segment encoding an antitermination protein and a 5765 bp DNA segment encoding the second set of stx 2d genes.
Production of Stx after exposure to antibiotics
The MIC of mitomycin C, enrofloxacin, and tetracycline was determined to be 6.25-, 0.08-, and 2.5 μg/mL, respectively; thus, a subinhibitory concentration of 0.5-, 0.05-, and 2.0 μg/mL, respectively, was used for Stxs induction. Production of Stx1a increased about 22.0- and 6.0-fold after exposure to mitomycin C and enrofloxacin, respectively (Fig. 5A); however, no change in Stx1a was observed for cells exposed to tetracycline (data not shown). Unexpectedly, unlike Stx1, the level of Stx2d was below the detection limit in both control cells and cells treated with any of the antibiotics described earlier, including mitomycin C (Fig. 5B).
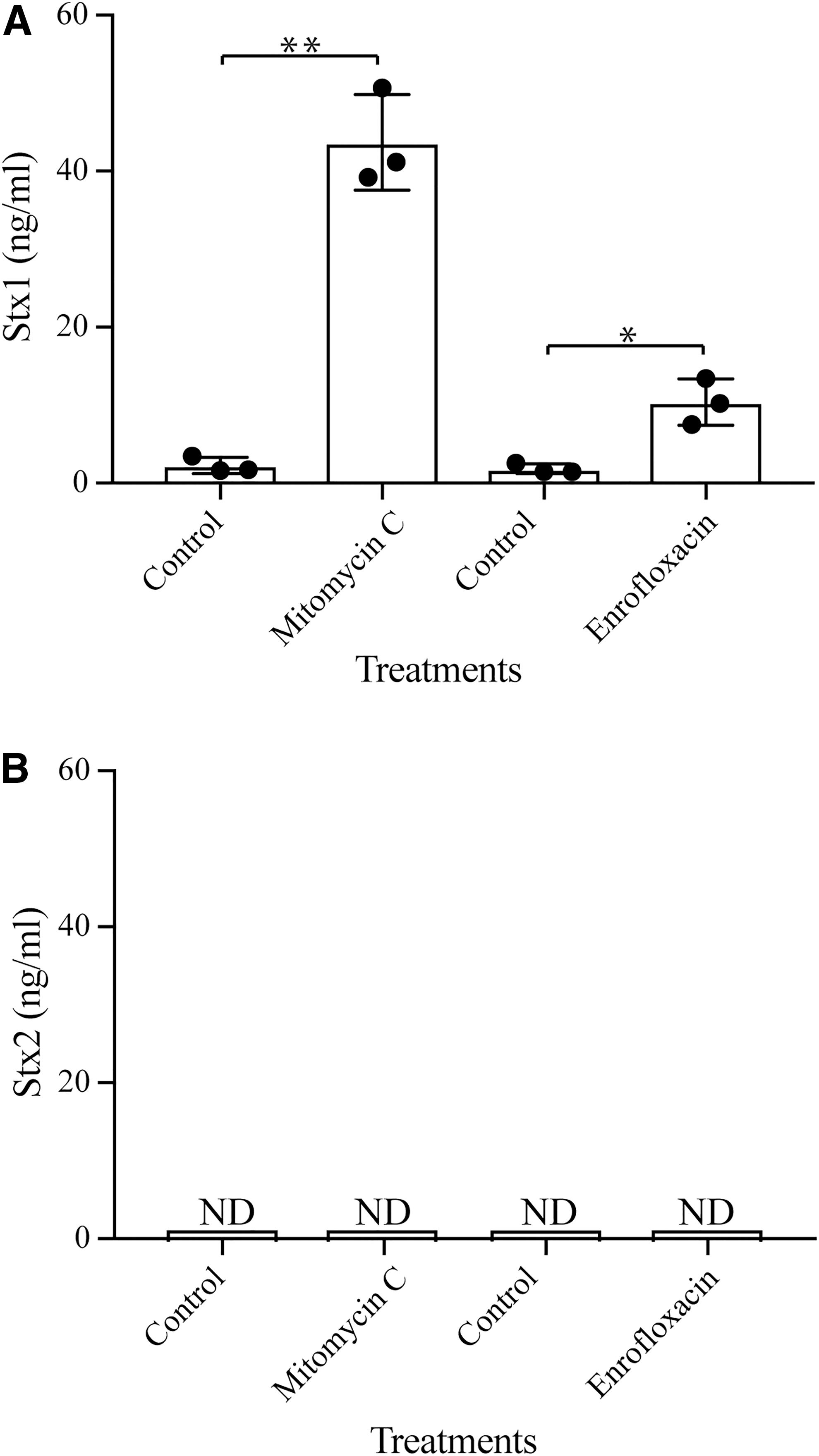
Production of Stxs in RM10809-C3 after antibiotic treatment.
Sequence analyses of stx and the P R′ region
The P R′ region and the CDSs of each stx in RM10809-C3 were compared with the corresponding DNA sequence in strain EDL933, a prototype of STEC. The CDS of stx 1a and the P R′ region are highly similar to that of stx 1a in EDL933 (Fig. 6A). Both the P R′ and Q protein utilization site, qut, are identical to those of EDL933. Further, the antitermination protein Q encoded by Stx1-prophage in RM10809-C3 shared 93.8% identity with that encoded by Stx1-prophage in EDL933 (Fig. 6A).
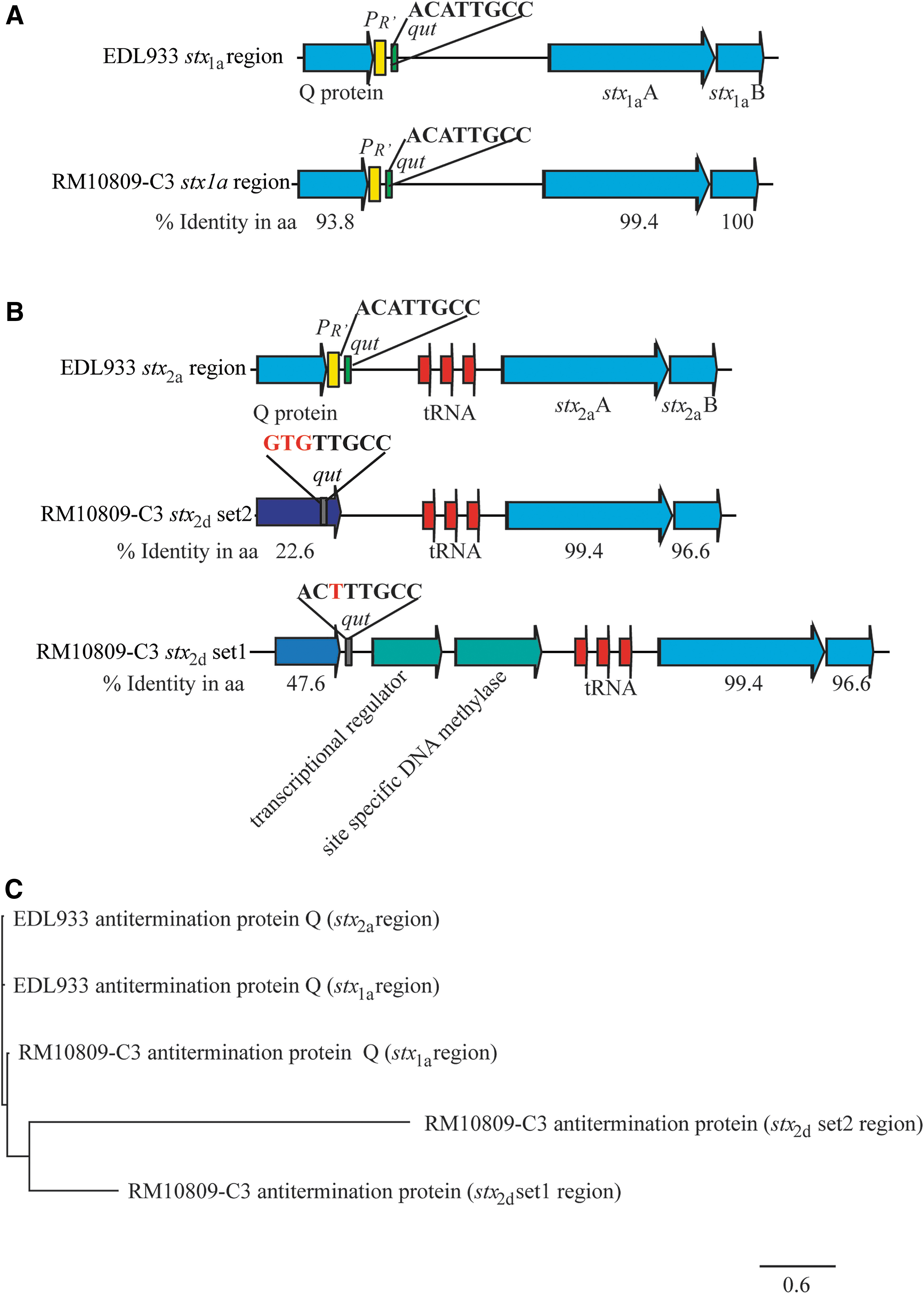
Sequence analyses of stx promoter region in RM10809-C3.
Although the CDS for both stx 2d are intact, large alterations were observed upstream of both stx 2d CDS (Fig. 6B). First, the classical lambda phage P R′ was not identified upstream of either stx 2d CDS. Second, the qut site was not conserved in any of the stx 2d promoter regions (Fig. 6B). Unlike the Q protein encoded by Stx1- or Stx2- prophage in EDL933, both antitermination proteins encoded by the Stx2d-prophage in RM10809-C3 were distantly related to the Q protein encoded by the Stx1-prophage in RM10809-C3, and to any of the Q proteins in EDL933 (Fig. 6C).
Discussion
To date, more than 500 STEC serotypes have been identified; however, only a subset is linked with human disease. The genetic basis of differences in STEC virulence remains unclear. Several molecular markers have been used to predict STEC pathogenicity potential. Strains positive in stx 2a and aggR, a plasmid-borne gene conferring cells aggregative phenotype (Beutin and Martin, 2012), are considered the highest risk groups, followed by stx 2a and eae in O157 and the “big six” serogroups. Strains carrying inducible Stx2d are also considered high risk, even if they do not possess eae, since cytotoxicity of Stx2d can increase significantly once activated by elastase (Melton-Celsa et al., 1996; Kokai-Kun et al., 2000). Our study revealed that, although strain RM10809-C3 possesses two intact CDS for activatable Stx2d, production of Stx2d was blocked. This natural silence of stx 2d is likely due to the alterations in the stx 2d promoters, including the qut cis element and the antitermination protein. Further examination upstream of stx 2d in other STEC genomes revealed vast sequence heterogenicity (Supplementary Fig. S2), which supports a previous report on the diverse stx promoters and their impact on stx expression (Zhang et al., 2018). Our data demonstrate that not only virulence gene content but also their regulatory components are critical for STEC risk assessment.
The two antitermination proteins encoded by RM10809-C3 Stx2-prophage are 126- and 273 aa in length, respectively, exhibiting 49.2% and 22.6% identity with the antitermination protein (144 aa) encoded by the RM10809-C3 Stx1-prophage, also known as Q933 (Plunkett et al., 1999). The 126-aa antitermination protein has been reported in sorbitol-fermenting O157 strains and in O111:H8 strain 11128, termed as QO111:H- (Haugum et al., 2012). The gene encoding QO111:H- is only detected in a small set of STEC strains and not always associated with stx. In contrast, the gene encoding the 273-aa antitermination protein (Q273) is widespread in STEC: It was detected in all STEC representative genomes examined in this study and in some strains, multiple copies were detected; however, it is not often associated with stx genes. Failure to detect Stx2d in both control and antibiotic-treated cells indicated that the molecular cues for de-repressing the QO111:H- or the Q273-controlled stx are likely different from those of the Q933. This finding supports a previous report that diverse P R′ regions are present in STEC and, among which, not all stx could be induced by mitomycin C (Zhang et al., 2018). Alternatively, the promoter strength downstream of QO111:H- or of Q273 might be much weaker than that of Q933, a similar control mechanism suggested for Q21-controlled stx genes (Koudelka et al., 2018).
The RM10809-C3 Stx2d-prophage is larger in genome size than any of the known Stx-converting phages. Blast search of NCBI nr database revealed parts of the Stx2d-prophage genome exhibiting high similarity with other STEC genomes, including O163:H19 str. FORC_41 (19,572 bp, 99.2% identity), O177:H25 strain 2014C-3011 (20,829 bp, 94.6% identity), O178:H19 strain 2012C-4431 (25,545 bp, 98.3% identity), and O137:H41 strain 88-3493 (39,310 bp, 99.6% identity). This mosaic structure indicates a robust recombination history in emerging Stx2d-prophage. In addition to genes encoding functions related to phage maturation, there are several genes encoding transporters, transcriptional regulators, and toxin-antitoxin systems, although their ecological contribution requires further investigation.
The complete genome sequence of RM10809-C3 enables us to assess its pathogenicity potential comprehensively as serotype O22:H8 is known to be associated with severe illness in humans. RM10809-C3 carries a complete LAA island, which would confer RM10809-C3 cells to attach and to colonize intestinal cells, which is an important step in pathogenesis of STEC, since the LAA was shown in STEC to contribute to colonization of the mice intestine (Montero et al., 2019). Our study presents an example of using whole genome sequences for risk assessment of STEC. Comparative genomics would lead us to discover the genetic basis of difference in virulence gene content among STEC strains as well as novel mechanisms governing expression of virulence traits. Such knowledge is valuable for rapid identification of emerging non-O157 STEC that may pose a serious public health risk.
Footnotes
Funding Information
This work was supported by the Current Research Information System (CRIS) of The United States Department of Agriculture (USDA)/The Agricultural Research Service (ARS) projects 2030-42000-050-00D and 2030-42000-049-00D.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.