
Abstract
Campylobacter jejuni is a major cause of bacterial gastroenteritis worldwide. In this study, we report the comparative genomic and functional characteristics of C. jejuni YH002 recently isolated from retail beef liver. Whole-genome sequencing and annotation of the strain revealed novel genetic features, including an integrated intact phage element, multiple antimicrobial resistance (AMR) genes, virulence factors, and a Phd-Doc type toxin–antitoxin (TA) system. Phenotypic tests of AMR showed that C. jejuni YH002 was resistant to amoxicillin and tetracycline, which correlates with the AMR genes found in the strain. Comparative analysis of cell motility at genotypic and phenotypic levels identified discernible patterns of amino acid changes, which could explain the variations of motility among C. jejuni strains. Together, these results provide important clues to the genetic mechanisms of AMR and cell motility in C. jejuni. The finding of a Phd-Doc TA system in the genome of C. jejuni YH002 is the first report of this TA system in Campylobacter spp.
Introduction
Campylobacter spp. are a leading cause of human gastrointestinal illness worldwide. In the United States, the organism affects more than 1.5 million people yearly (
The ability of the organism to cause human infections is attributed to a range of virulence properties, namely motility, adhesion, invasion, toxin production, iron uptake, and antimicrobial resistance (AMR). Although some of these virulence factors have been studied in considerable detail, understanding of the disease-causing mechanisms is still far from complete (Dasti et al., 2010; Bolton et al., 2015). Of particular interest is the flagellar motility, which is essential for C. jejuni survival, colonization, and establishment of a successful infection in the host intestinal tract. Many genes are believed to be associated with the motility and chemotaxis of C. jejuni and novel motility-related genes are still reported (Guerry, 2007; Gao et al., 2014).
Another area of concern is the emergence of AMR in Campylobacter spp. The World Health Organization recently listed Campylobacter spp. as a high-priority organism for an urgent need to address AMR (
With advancements in and application of whole-genome sequencing (WGS), the studies of foodborne pathogens have gained multiple benefits complementing traditional microbiological methods, including successful prediction of AMR and virulence properties. Though researchers sequenced several C. jejuni genomes, available in public databases, there are very few comparative genomic studies on C. jejuni food isolates (Skarp et al., 2015; Zhao et al., 2016). In this study, we isolated and characterized a C. jejuni strain YH002 from retail beef liver. Multidrug resistance and high motility of the strain suggested that it was a potentially disease-causing agent, which could greatly threaten food safety and public health. Therefore, we report the comparative analysis of the genome and phenotypes of C. jejuni YH002 with respect to the well-characterized C. jejuni strains 81-176 (Hofreuter et al., 2006) and NCTC 11168 (Parkhill et al., 2000) to provide a better understanding of the genetic diversity and pathogenicity of this important foodborne pathogen.
Materials and Methods
Bacterial strains, isolation and identification
C. jejuni YH002 was isolated from retail beef liver by using a passive filtration technique (He et al., 2015). Briefly, 450 g of liver was rinsed with 200 mL of 0.1% buffered peptone water (Difco, Becton Dickinson, NJ) followed by concentration and overnight enrichment in Bolton broth (Difco, Becton Dickinson) containing horse blood (Remel, Thermo Fisher Scientific, NJ) and antibiotic supplements. The enriched broth (100 μL) was pipetted onto a 0.65-μm membrane (Millipore, Billerica, MA) on a Brucella agar plate for 15 min to allow Campylobacter cells to pass through. The membrane was then removed, and plates were incubated for 24 h under microaerophilic conditions (5% O2, 10% CO2, and 85% N2) at 42°C. The strain was confirmed by a real-time PCR assay targeting hipO (He et al., 2010).
WGS, assembly and annotation
Genomic DNA was purified by using the Qiagen Genomic-tip 100/G kit (Qiagen, Valencia, CA) and quantified by Qubit 3.0 fluorometer (Thermo Fisher Scientific, Waltham, MA). To obtain high-quality WGS data, both Single Molecule Real-Time (Pacific Bioscience, Menlo Park, CA) and MiSeq sequencings (Illumina, San Diego, CA) were undertaken (Ghatak et al., 2017). PacBio and Illumina reads were de novo assembled by using Canu v1.3 (Koren et al., 2017) and Spades v3.7.1 (Bankevich et al., 2012), respectively. The contigs from both assemblies were unified and corrected by Pilon (
Motility assay
The motility of C. jejuni YH002, 81-176, and NCTC 11168 was evaluated on 0.4% Muller Hinton (MH) agar plates (Difco, Becton Dickinson) as previously described (Golden and Acheson, 2002). Strains were grown in MH broth and then diluted to an OD600 = 0.15. Adjusted cultures (2 μL) were spotted onto 0.4% MH plates with a uniform agar thickness. After incubation at 37°C for 48 h, diameters of the halo zones were measured and photographed with a Molecular Imager Gel Doc (Bio Rad Laboratories, Hercules, CA) under “epi-white” light.
Antimicrobial susceptibility test
The minimum inhibitory concentrations (MICs) of nine antimicrobials (Table 1) were determined for C. jejuni YH002, 81-176, and NCTC 11168 by the broth micro-dilution technique as per Clinical Laboratory Standards Institute (2015) guidelines by using Sensititre Plates (ThermoFisher Scientific, Cleveland, OH). C. jejuni ATCC 33560 was included as a control. In addition, MICs were determined for four antimicrobials (streptomycin, amoxicillin, ertapenem, and polymyxin) by using ETEST MIC strips (Biomerieux, Boston, MA) as per the manufacturer's instructions. Results were interpreted by using epidemiological cut-off values provided in NARMS (
Antimicrobial Susceptibility and Occurrences of Antimicrobial Resistance Genes in Campylobacter jejuni Strains
The control strain C. jejuni ATCC 33560 was sensitive to all the antibiotics tested except for Polymyxin B.
Putative efflux pumps related genes are not included, except for macA and macB, which are reported to be antibiotic class specific.
GEN, gentamicin; STM, streptomycin; AMX, amoxicillin; ERT, ertapenem; TEL, telithromycin; CLI, clindamycin; AZI, azithromycin; ERY, erythromycin; FLO, florfenicol; POL, polymyxin B; CIP, ciprofloxacin; NAL, nalidixic acid; TET, tetracycline; MIC, minimum inhibitory concentration.
In silico analyses of phage, AMR genes, motility-associated genes, and multilocus sequence type
The genome of C. jejuni YH002 was analyzed for phage insertion by using PHASTER (Arndt et al., 2016) and searched for AMR genes in the Comprehensive Antibiotic Resistance Database (CARD) (Jia et al., 2017). Chemotaxis- and motility-related sequences were extracted from all three genomes, and the translated amino acid sequences were aligned by MUSCLE (Edgar, 2004). Alignments were visualized by Jalview (Waterhouse et al., 2009). Differences in the amino acids were identified, tabulated, and analyzed by using Microsoft Excel. BLAST comparison of the genomic regions for motility-associated genes was performed, and figures were drawn with Easyfig (Sullivan et al., 2011). Multilocus sequence typing of the genomes was performed by using Campylobacter multilocus sequence type (MLST) server (
Results
Genome of C. jejuni YH002: assembly and annotation
The strain YH002 was isolated from retail beef liver and identified by the real-time PCR. WGS yielded a complete genome of 1,820,488 bp (30.4% GC content), comprising a 1,774,584 bp chromosome (GenBank accession No. CP020776) and a 45,904 bp plasmid pCJP002 (CP020775). pCJP002 contained tetO and a type IV secretion system with a sequence identity of 99.81% and 99.41% to pAR-0415 (CP044168.1) and pTet-M129 (CP007750.1), respectively.
RAST annotation of the YH002 genome predicted 1928 coding sequences (CDSs) with 53 RNAs. By the same approach, C. jejuni 81-176 and NCTC 11168 yielded comparatively fewer CDSs (1751 and 1689, respectively). There were 77 genes associated with virulence, disease causation, and defense mechanisms in YH002, more than the other two strains. Interestingly, four phage-related elements found in YH002 were completely absent in 81-176 and NCTC 11168.
Comparative genomic map (Fig. 1) showed that YH002 had a larger genome than 81-176 and NCTC 11168 as evident from two large gaps in the latter two genomes at 1220–1310 kb and 1410–1445 kb. Although the gap at 1410–1445 kb was occupied by an integrated phage element, the other region (1220–1310 kb) harbored a number of genes unique to C. jejuni YH002. In addition, the genome carried various functional genes related to AMR, restriction modification systems, pathogenicity island protein (Cag12), cytolethal distending toxin (Cdt), toxin–antitoxin (TA) system (Phd-Doc), and CRISPR (Cas1, Cas2, and Csn1).

Comparative genome map of Campylobacter jejuni YH002, 81-176, and NCTC 11168. Selected features of the C. jejuni YH002 genome are indicated in the outermost ring.
MLST analysis indicated that C. jejuni YH002 and NCTC 11168 belonged to the same clonal complex (CC ST-21), whereas C. jejuni 81-176 was classified into CC ST-42. Sub-categorization of the genomes, however, separated them into three different sequence types (C. jejuni YH002 to ST982, C. jejuni NCTC 11168 to ST43, and C. jejuni 81-176 to ST604).
Integrated phage in the genome of C. jejuni YH002
RAST annotation indicated the presence of an integrated phage element in the chromosome of C. jejuni YH002. This prediction was confirmed by searching against the PHASTER database, which identified a 45.9 kb intact phage located between 1.4 and 1.44 Mb (Fig. 2). It shares 100% sequence identity with the prophage CJIE1 in C. jejuni 00-2425, 00-2538, 00-2544, and RM3420 by BLAST against all the Campylobacter genomes in GenBank at National Center for Biotechnology Information. With 30.36% GC content, this phage element had two attachment sites along with the necessary portal, shaft, fiber, and plate proteins as well as integrase. The PHASTER query yielded a score of 150 for the element, indicating an intact phage. In addition, there were other phage-associated genes in the genome, including two copies of genes encoding phage Rha proteins and phage antirepressors.
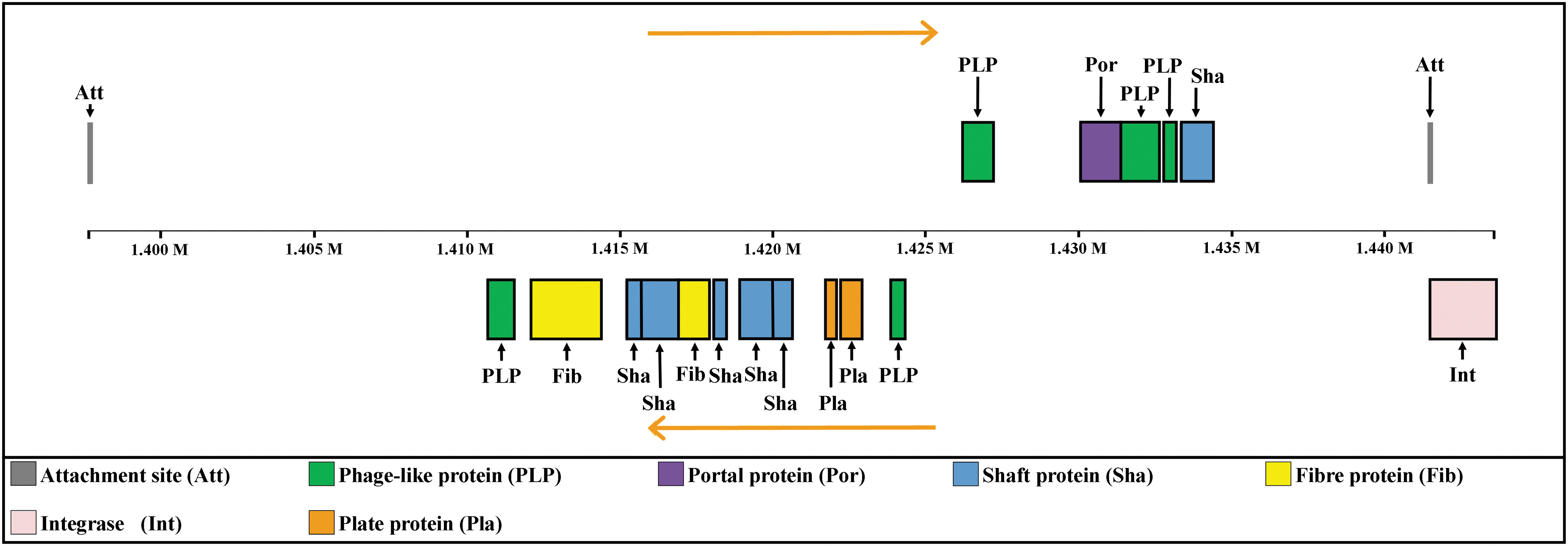
A genetic map of a phage integrated in the genome of C. jejuni YH002 as identified by PHASTER.
Motility
Motility of C. jejuni YH002, 81-176, and NCTC 11186 was compared at the genetic and phenotypic levels. Motility- and chemotaxis-associated genes were distributed over large regions in all the genomes. However, BLAST alignment of these sequences indicated an inversion (red) and two insertions (highlighted in gray) of DNA segments in YH002 (Fig. 3a). A closer comparison of several motility genes revealed divergent genomic rearrangements, with an inserted sequence in NCTC 11168 along with a truncated gene.

Comparative analysis of the motility of C. jejuni YH002, 81-176, and NCTC 11168.
A phenotypic test of motility revealed that C. jejuni 81-176 was the most motile followed by YH002 and NCTC 11168 with a ratio of halo diameters of 6.5:1.9:1.0 (Fig. 3b).
RAST predicted numerous motility- and chemotaxis-associated genes in the C. jejuni genomes (82 in 81-176, 86 in NCTC 11168, and 85 in YH002). Amino acid sequence alignments of all these genes indicated many amino acid substitutions occurring in numerous genes. Figure 3c delineates the relative number of amino acid variations between the strains as revealed by the alignments. The most motile strain (C. jejuni 81-176) had the most unique amino acid sequences in the motility and chemotaxis genes, carrying 51.8% of the observed single amino acid changes (replacements/insertions/deletions). By comparison, strains NCTC 11168 and YH002 contained 16.3% and 10.3% of the unique amino acid residues whereas 21.7% of the variable residues were different in all three strains. The distinct sequence differences of the motility-associated genes in 81-176 possibly resulted in higher motility of this strain.
Since flagella-driven motility in bacteria involves multiple genes, we further investigated the CDSs affected by amino acid changes across the entire flagellar apparatus. Figure 3d shows that the affected genes (labeled in red) were not restricted to certain functional groups. Instead, they were distributed in various sub-components, including motor switches, C ring, MS ring, P ring, proximal and distal rods, hook length control region, hook cap, and motility accessory factors.
Antimicrobial resistance
Antimicrobial susceptibility tests showed that YH002 had substantially higher MICs to amoxicillin (64 μg/mL) and tetracycline (32 μg/mL) than other two strains (Table 1). On RAST annotation and CARD query, we analyzed the putative resistance genes from the three genomes. Interestingly, bla OXA-61 (encoding beta-lactamase) was found in both YH002 and NCTC 11168, but the MIC of NCTC 11168 (1.5 μg/mL) to amoxicillin was 42-fold lower than YH002 (64 μg/mL). Sequence alignment (Fig. 4a) revealed a point mutation (G → T) in the promoter of bla OXA-61 in YH002, which presumably restored the TATA box, improved the expression of the gene, and, therefore, increased the resistance to amoxicillin.

The tetO gene, which encodes a protein that protects the ribosome from binding tetracycline, was observed in C. jejuni YH002 and reported in C. jejuni 81-176. However, strain 81-176 was susceptible to tetracycline with an MIC (0.25 μg/mL) 128-fold lower than C. jejuni YH002 (32 μg/mL). Genomic analysis found that YH002 harbored two copies of tetO (one in chromosome and another in plasmid), whereas 81-176 only contains a plasmid copy of the gene that could be spontaneously lost during cell growth (Bacon, 2000). Real-time PCR using the primers specific to tetO (Aminov et al., 2001) confirmed the instability of the plasmid-carrying gene in 81-176 in the absence of antibiotic selection. Sequence alignment of TetO revealed eight instances of amino acid replacement and a shorter CDS of the plasmid copy in YH002 (Fig. 4b), which also could contribute to the tetracycline resistance in the strains.
Despite the presence of the genes for macrolide-specific efflux pumps (macA, macB) and mfd, which promote spontaneous mutations in the fluoroquinolone target, there was no corresponding resistance to macrolides (azithromycin, erythromycin) or to ciprofloxacin in any of the C. jejuni strains. Similarly, the presence of aac-(3) and pmrE in these strains did not confer the resistance to gentamycin and polymyxin B, respectively.
In addition to the genes described in Table 1, all the strains harbored genes encoding resistance to aminocoumarin antibiotics (alaS) and tiamulin (taeA), and an array of efflux pump genes including cmeABCR, multidrug efflux transporter, and multi-antimicrobial extrusion proteins.
Discussion
High prevalence of Campylobacter spp. in retail beef liver and enhanced survivability in liver juices have been reported (Noormohamed and Fakhr, 2013; Karki et al., 2019). In this study, we isolated C. jejuni strain YH002 from the enrichment of beef liver rinse. However, it does not exclude the origin of the strain from other parts of the organs or intestine due to the cross-contamination in meat processing.
WGS and annotation revealed a 45.9 kb intact phage element in the chromosome of C. jejuni YH002, suggesting that a substantial portion of its genetic material was horizontally acquired. Phage integration into the genome of C. jejuni and its influence on gut colonization and formation/dispersal of biofilms were previously reported (Scott et al., 2007; Clark et al., 2014; Johnson et al., 2017; Siringan et al., 2014). Because the YH002 strain harbors an intact phage, it assumes greater importance in attachment/infection and rapid dispersal of biofilms triggered by phage reproduction.
An interesting feature found in C. jejuni YH002 was a TA system comprising a phd (prevent-host-death) and two copies of doc (death-on-curing) in the chromosome. TA systems in bacteria, especially those located in plasmids, are known to maintain plasmid stability by post-segregational killing of plasmid-free cells. However, the physiological roles of chromosome-encoded TA systems differ from their plasmid counterparts and are believed to be involved in stress response, biofilm formation, and persistence (Page and Peti, 2016). Among the five major types of TA systems in bacteria, the type II TA system, comprising virA/virT, has been recently characterized in the C. jejuni plasmid (Van Melderen and Saavedra De Bast, 2009; Shen et al., 2016). In this study, we report the first identification of the Phd-Doc system in Campylobacter spp. The presence of a chromosomal Phd-Doc system in YH002 could possibly stabilize neighboring genes and integrated genetic elements (i.e., bacteriophages) as demonstrated in other microorganisms (Guerout et al., 2013).
Motility, a key virulence factor in C. jejuni, is intricate and involves the interplay of a number of genes associated with chemotaxis and flagella. Previous studies showed that the disruption of any component in flagellar apparatus or chemosensory pathways of C. jejuni has a significant effect on its host colonization (Lertsethtakarn et al., 2011; Korolik, 2019). Our results demonstrated that C. jejuni 81-176 was highly motile compared with the other two strains, which was not surprising because 81-176 is a known outbreak strain. Single amino acid replacements, insertions, and deletions, which were most frequent in 81-176, could possibly be the genetic cause of its increased motility/pathogenicity. Moreover, MLST showed that 81-176 (ST-42) is genetically divergent from YH002 and NCTC 11168, both of which are in ST-21. Interestingly, we found that amino acid changes affected multiple components of the flagellar apparatus. These genetic variations did not completely halt the motility of C. jejuni strains, highlighting the inherent versatility of the flagellar system.
The results of antimicrobial susceptibility tests showed that C. jejuni YH002 was resistant to tetracycline and amoxicillin. Despite possession of bla OXA-61 in both YH002 and NCTC 11168, an elevated MIC (>42-fold) in YH002 is likely due to the point mutation (G → T) in the TATA box of bla OXA-61 in this strain (believed to increase the gene expression). This result agrees with the previous finding (Zeng et al., 2014).
Tetracycline has been used in animal production for many years and high prevalence (88%) of tetracycline resistance was recently reported in 320 C. jejuni isolates from cattle (Tang et al., 2017). Hence, it is not surprising that C. jejuni YH002, a beef isolate, was resistant to tetracycline with an MIC (32 μg/mL) 128-fold higher than the other two strains. An extra copy of tetO in the chromosome of YH002 in addition to its plasmid version might account for a high level of tetracycline resistance of the strain. Moreover, eight amino acid changes were noticed at various locations of tetO in these strains. Whether these genetic changes solely accounted for substantial MIC difference remains speculative, but the tetracycline resistance genotype and phenotype of YH002 did correspond to previously reported resistance patterns in C. jejuni (Zhao et al., 2016).
Campylobacter spp. are inherently resistant to polymyxin B due to the lack of an appropriate target (Iovine, 2013), as was also evidenced in our study (Table 1). Lastly, our genomic and phenotypic data could not distinctively infer the roles of efflux pumps in mediating AMR in the organism, though efflux systems have been reported to often act as synergistic components with other mechanisms of resistance rather than operating independently in Campylobacter spp. (Luangtongkum et al., 2009; Yao et al., 2016; Vieira et al., 2017).
Conclusions
Taken together, our results provide important clues to the genetic mechanisms of AMR and motility of C. jejuni. Novel outcomes revealed in the study include an integrated intact phage, multiple AMR genes and virulence factors, and a phd-doc TA system in C. jejuni YH002. Resistances of the strain to tetracycline and amoxicillin correlated with the presence of and/or variations in associated genes. Altered motility of the strain was perhaps driven by discernible patterns of amino acid changes in the flagellar apparatus. Existence of the phd-doc TA system is the first instance to be reported for Campylobacter spp.
Footnotes
Acknowledgments
The first author (S.G.) is thankful to the Department of Biotechnology, New Delhi, India for Overseas Associateship Grant for NER (2015–2016) and to the Indian Council of Agricultural Research for necessary support. The authors thank Dr. Patricia Guerry, Naval Medical Research Center (Silver Spring, MD), for providing C. jejuni strain 81-176.
Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by the United States Department of Agriculture, Agricultural Research Service with the Current Research Information System number 8072-42000-084.