
Abstract
Klebsiella pneumoniae is considered an opportunistic pathogen, constituting an ongoing health concern for immunocompromised patients, the elderly, and neonates. Reports on the isolation of K. pneumoniae from other sources are increasing, many of which express multidrug-resistant (MDR) phenotypes. Three phylogroups were identified based on nucleotide differences. Niche environments, including plants, animals, and humans appear to be colonized by different phylogroups, among which KpI (K. pneumoniae) is commonly associated with human infection. Infections with K. pneumoniae can be transmitted through contaminated food or water and can be associated with community-acquired infections or between persons and animals involved in hospital-acquired infections. Increasing reports are describing detections along the food chain, suggesting the possibility exists that this could be a hitherto unexplored reservoir for this opportunistic bacterial pathogen. Expression of MDR phenotypes elaborated by these bacteria is due to the nature of various plasmids carrying antimicrobial resistance (AMR)-encoding genes, and is a challenge to animal, environmental, and human health alike. Raman spectroscopy has the potential to provide for the rapid identification and screening of antimicrobial susceptibility of Klebsiella isolates. Moreover, hypervirulent isolates linked with extraintestinal infections express phenotypes that may support their niche adaptation. In this review, the prevalence, reservoirs, AMR, Raman spectroscopy detection, and pathogenicity of K. pneumoniae are summarized and various extraintestinal infection pathways are further narrated to extend our understanding of its adaptation and survival ability in reservoirs, and associated disease risks.
Introduction
K
Interactions between humans, animals, and the environment can have an impact on public health, as a source of diseases. According to the World Health Organization (WHO) definition, diseases, naturally transmissible from animals to humans through direct contact or food, water, and the environment, are commonly referred to as zoonoses. At least 61% of all human pathogens are zoonotic, and zoonoses comprise a large percentage (75%) of all newly identified or emerging infectious diseases as well as existing infectious diseases during the past decade. With the exception of several newly emerging zoonoses such as SARS and influenza H5N1, most are not prioritized by health systems at national and international levels, and are therefore labeled as neglected. Although not listed in the WHO zoonoses inventory, K. pneumoniae was reported as a zoonotic disease in several publications (Whitehouse et al., 2010; Soto et al., 2012; Zhang et al., 2019; Santaniello et al., 2020).
Taxonomy and Phylogroups of K. pneumoniae
Based on the sequence of several genes and whole-genome sequencing data of more than 300 human and animal isolates from four continents, K. pneumoniae can be divided into three distinct phylogroups: these comprise K. pneumoniae (KpI) (Fevre et al., 2005), K. quasipneumoniae (KpII A-B) (Brisse et al., 2014), and K. variicola (KPIII) (Rosenblueth et al., 2004; Maatallah et al., 2014), although all are able to colonize and infect humans, KpI is frequently associated with human infection (Holt et al., 2015).
Among these three phylogroups, a pangenome of 29,886 unique protein-coding sequences can be detected. Furthermore, among 328 K. pneumoniae genomes studied, these shared ∼1900 core genes, giving a median of 5705 genes per genome. The remaining genes are considered to be accessory and more than 65% of these are considered rare. Some of these accessory genes are likely to be localized on plasmids acquired from a wide range of bacteria, including other members of Enterobacteriaceae, Vibrio species, and Acinetobacter species (Holt et al., 2015). Genomic plasticity along with its ability to exchange plasmids with other species makes this bacterium an important reservoir of antimicrobial resistance (AMR) and virulence genes (Ramirez et al., 2014).
To gain a better understanding of the biology of this clinically important bacterium, further large-scale comparative and functional genomics studies are required (Wyres and Holt, 2016).
Based on a recent review, the systematics of K. pneumoniae was updated by performing population genomics, and a whole genome-based tree was generated from representative complete genome sequences to show the phylogenetic relationships between the K. pneumoniae species complex (KpSC), other select members of the Klebsiella genus, and family Enterobacteriaceae, and to advance our understanding of K. pneumoniae taxonomy, evolution, diversity, and distribution (Wyres et al., 2020). From this phylogenetic tree, five species and/or seven phylogroups were included in the K. pneumoniae complex (“Kp”) with KpII and KpIII being split into two subspecies: K. pneumoniae sensu stricto (Kp1), K. quasipneumoniae subsp. quasipneumoniae (Kp2) (Brisse et al., 2014), K. quasipneumoniae subsp. similipneumoniae (Kp4) (Brisse et al., 2014), K. variicola subsp. variicola (Kp3) (Rosenblueth et al., 2004), K. variicola subsp. tropica (Kp5) (Rodrigues et al., 2019), K. quasivariicola (Kp6) (Long et al., 2017), and K. africana (Kp7) (Rodrigues et al.,
Epidemiology of Klebsiella
Human diseases caused by Klebsiella
Despite being benign in many instances, this bacterium is often linked with community-acquired (CA) or hospital-acquired (HA) infections. HA K. pneumoniae are responsible for bloodstream infections, along with infections of the abdominal cavity, surgical sites, soft tissues, meningitis, and respiratory and urinary tracts, particularly in immunocompromised patients, the elderly and neonates (Podschun and Ullmann, 1998; Lee and Burgess, 2012; Janda, 2015). The most common site of infection is the blood (52%), followed by the respiratory tract (30%), and urine (10%) (Lee et al., 2012). However, since the 1980s, a new more virulent variant of Klebsiella has been disseminating globally, being responsible for CA K. pneumoniae. This strain was first described in the Asian Pacific Rim and has now spread globally (Pastagia et al., 2008; Sobirk et al., 2010; Decré et al., 2011; Fierer et al., 2011; Vila et al., 2011). This variant is known for its hypervirulent (hypermucoviscous) phenotype and for the presence of specific capsule serotypes K1 and K2, along with various siderophores (Shon et al., 2013). This bacterium is capable of causing pyogenic liver abscesses (PLAs), Lemierre's syndrome, and antibiotic-associated hemorrhagic colitis, and has a propensity to migrate to other parts of the body with mortality rates ranging from 5% up to 30% (Janda, 2015).
Molecular methods for subtyping Klebsiella isolates
There are several molecular methods that can be used for subtyping K. pneumoniae. These include repetitive sequence-based PCR, pulsed-field gel electrophoresis, multilocus sequence typing (MLST), and whole-genome sequencing. MLST is the most commonly deployed method to identify K. pneumoniae and is based on genetic variation detected among seven housekeeping genes (rpoB, gapA, mdh, pgi, phoE, infB, and tonB). The sequence type (ST) is determined based on the different allelic numbers assigned to each distinct sequence allele within each locus (Diancourt et al., 2005). The ST numbers are then combined into closely related groups to form clonal complexes (CCs) (Turner et al., 2007). Recently a new approach proposed to group these CCs into clonal groups (CGs). In this study, the most predominant STs would be considered the center of the CG and include both single-locus variants and their respective variants (Breurec et al., 2013). This approach would allow the identification of epidemiologically related strains to provide more meaningful information regarding their clonal evolution to the clinicians.
Epidemic Klebsiella CGs
Epidemic K. pneumoniae are restricted to specific clonal lineages (Lai et al., 2019). HA infections are known to be caused by MDR strains, especially those that are resistant to carbapenem-type compounds, primarily by acquiring the carbapenemase-encoding genes on mobile genetic elements (MGEs). These isolates normally belong to the CG258, comprising the ST258 and ST512 types (Bowers et al., 2015; Kelly et al., 2019; Palmeiro et al., 2019; Lai et al., 2019). In contrast, CA infections are known for their extreme virulence and limited resistance to antimicrobial agents and are normally associated with the CG23 group (Passet and Brisse, 2015; Lam et al., 2018; Lai et al., 2019), along with other CG groups, including CG65 and CG86 (Brisse et al., 2009; Decré et al., 2011; Passet and Brisse., 2015). However, several exceptions have been noted and it has been suggested that the evolution of K. pneumoniae could be due to homologous recombination rather than the accumulation of mutations (Brisse et al., 2009). The presence of hypervirulent and antimicrobial-resistant K. pneumoniae in food matrices poses a potential hazard to public health (Zhang et al., 2018). Although K. pneumoniae populations carry both drug resistance and hypervirulence markers and are found to be largely nonoverlapping in BIGSdb-Kp, a database that could enable rapid extraction of medical and epidemiological information from K. pneumoniae genomes, isolates with these features are still being detected (Bialek-Davenet et al., 2014).
Source of infection
Klebsiella is considered an opportunistic pathogen that can be found in the environment (Seidler et al., 1975; Barati et al., 2016), and in the gastrointestinal (GI) tracts of humans and animals (Bagley, 1985; Kim et al., 2005). It has the ability to adapt to an oxygenated or nonoxygenated environment (Keynan and Rubinstein, 2007). It is also found on the skin, and it can be cultured from the nose and throat of healthy individuals as a benign colonizer (Imhoff, 2005; Brisse et al., 2006; Berrazeg et al., 2013). In terms of Klebsiella of food origin, the following section describes the prevalence and reservoirs in this context.
Prevalence and Reservoirs of Foodborne Klebsiella species
Klebsiella can be found in different environments depending on the phylogroup. K. pneumoniae (KpI) is commonly associated with mammalian infection and can be found in hospital- and community-based environments. MDR and virulent CGs, CG258 and CG23, belong to this phylogroup. K. quasipneumoniae (KpII) can also be found in the same settings as KpI; however, this phylogroup does not demonstrate the same virulence capacity as K. pneumoniae, acting more as a carrier than a pathogen (Holt et al., 2015), nonetheless the primary reservoir remains unknown. In contrast, KpIII, K. variicola, has a specific environmental niche. This bacterium is commonly associated with plant-based infections (Rosenblueth et al., 2004; Wei et al., 2014; Martínez-Romero et al., 2015; Zurfluh et al., 2015). Ultimately, all three pathogenic species can cause human infections.
It had been recognized for a long period that hospital inpatients are more likely to be intestinal carriers of Klebsiella when compared with the nonhospital population, a feature that may probably be related to antimicrobial therapy (Rose and Schreier, 1968). Furthermore, until recently, K. pneumoniae was described as a human pathogen with attention being focused on related nosocomial infections (Campos et al., 2016). Recently, foodborne outbreaks associated with the Klebsiella genus have been reported with increasing numbers in different countries (Zhang et al., 2018). Similarly, gastroenteritis associated with contaminated turkey occurred at a catered company meal (Rennie et al., 1990). K. pneumonia, from a contaminated hamburger, was reported officially as an enteroinvasive foodborne pathogen for the first time in 1998 (Sabota et al., 1998); in this case, a previously healthy male ingested a portion of a hamburger from a fast food chain, after which, symptoms of gastroenteritis quickly developed into multiorgan failure. This report highlighted a possible hitherto unrecognized environmental niche for Klebsiella and that had not previously been reported as enteroinvasive.
The prevalence of Klebsiella species has been identified in various food products around the world and the food categories vary, consisting of turkey, pork, cake, cattle, chicken, milk, retail meat product, hamburger, fish and prawn, and even infant formula milk powder. It was reported as a frequent isolate among Enterobacteriaceae cultured from meats and meat-handling surfaces in packing plants. Occurrence reports of K. pneumonia in street food samples were also reported from different states in Malaysia (Haryani et al., 2007) and Benin (Moussé et al., 2016). The category details of Klebsiella recovered from foods are shown in Table 1.
Food Categories of Klebsiella Recovered
RTE, ready-to-eat.
Apart from clinical origins, it appears that this pathogen can be recovered from various food sources, a feature overlooked due to the absence of surveillance. Moreover, a foodborne nosocomial outbreak due to extended-spectrum β-lactamase (ESBL)-producing K. pneumoniae in a hospital setting in Spain was reported, which prompted an epidemiological investigation of a possible foodborne transmission of KpI (Calbo et al., 2011). In this case, 156 patients were colonized or infected, more than one-third of hospital kitchen—screened surfaces or foodstuffs were colonized and 15% of food handlers were found to be fecal carriers.
The transmission of Klebsiella species has also been reported from cattle farms and its dissemination is mediated by biting midges and houseflies (Mohammed et al., 2016). The bacterium may also be found contaminating vegetables fertilized with untreated wastewater (Ibenyassine et al., 2007), and in samples from pig farms (García-Cobos et al., 2015), tap and pond water (Babu et al., 2015), natural surface water of the Baltic Sea (Podschun et al., 2001), milk samples from dairy farms (Amosun et al., 2012; Sudarwanto et al., 2015), and air samples taken from broiler houses (Bródka et al., 2012). K. pneumoniae and K. variicola can also cause bovine mastitis (Kanevsky-Mullarky et al., 2014; Osman et al., 2014; Podder et al., 2014). The same pathogens were also found in cow's milk as part of the microbial community in subclinical mastitis cases (Locatelli et al., 2010; Bhatt et al., 2012; Saishu et al., 2014; Timofte et al., 2014; Ali Hassan Ahmed Al-Shammary, 2017).
The poor characterization of the STs identified from most of these studies hampers the identification of the bacterial reservoirs and provides little insight into how these bacteria may be associated with cases of human infection. Nonetheless, others reported a close phylogenetic relationship between clinical isolates and those cultured from retail sources (Davis et al., 2015; Hartantyo et al., 2020), bovine mastitis (Holt et al., 2015), livestock (Davis and Price, 2016), and food-producing animals (Wang et al., 2018). Moreover, it was reported that meat sources were more prone to be contaminated with MDR Klebsiella species when compared with clinical isolates (Davis et al., 2015), a feature that could be due to the use of antimicrobial agents in food-animal production. The loss of the MDR phenotype in clinical isolates could be due to the adaptation of bacteria to the new environment, losing nonessential genes that could affect the metabolism of the bacterium.
Once these hypervirulent phylogenetic lineages have been identified in the environment (Osman et al., 2014; Overdevest et al., 2014; Davis et al., 2015; Holt et al., 2015), it facilitates our ability to predict how cases of community-hypervirulent Klebsiella infections will emerge over the coming years.
Klebsiella species can be detected in the environment, especially in animal and plant production settings, and these could be a hitherto unexplored reservoir. Intercountry passage of opportunistic Klebsiella as contaminants of fresh produce and meats could have been one of the reasons for the dissemination of this pathogen, especially given the fact that no food safety surveillance is legally mandated for this bacterium (Zurfluh et al., 2015; Al-Kharousi et al., 2016).
AMR Detected in Klebsiella species
Several members of the Enterobacteriaceae family have the capacity for carrying and transmitting a wide variety of resistance-encoding genes through horizontal gene transfer. One of the areas of most concern, in respect of human health, is the rapid dissemination of AMR among K. pneumonia. Some of the major resistance mechanisms for K. pneumoniae are summarized in Table 2. In K. pneumoniae, MDR evolution is largely driven through the acquisition of AMR genes on diverse mobilizable plasmids (Navon-Venezia et al., 2017), such as CG 258, which is implicated in global spread of the K. pneumoniae carbapenemases (KPCs) (Chen et al., 2014b). In 2017, the etiological agent responsible for a fatal outbreak was a carbapenem-resistant K. pneumoniae belonging to CG258, which had acquired ICEKp in the chromosome plus iuc and rmpA2-encoding genes on a virulence plasmid (Gu et al., 2018).
Antimicrobial Resistance Genes or Mutations in Klebsiella pneumoniae
ESBL, extended-spectrum β-lactamase.
K. pneumoniae is known for its diversity of AMR elements and was described as a key trafficker of drug resistance genes from the environment to the clinical. This is mainly due to its ability to co-transfer AMR genes arising from its wider ecological distribution, its significantly varied DNA composition, its greater AMR gene diversity, and a higher plasmid burden when compared with other Gram-negative opportunists (Wyres and Holt, 2018). The diversity in gene content and context found in the Klebsiella genome is mainly due to plasmids, bacteriophages, insertion sequence, and integrative conjugative elements (ICEs) (Holt et al., 2015; Mathers et al., 2015).
During the 1980s and 2000s, K. pneumoniae was recognized for carrying plasmids that encode resistance mechanisms to counter aminoglycoside, fluoroquinolone, and ESBL classes of compound. The latter are mostly composed of TEM-, AmpC-, CTX-, OXA-, and SHV-type enzymes (Soilleux et al., 1996; Brisse et al., 2000; Li and Lim, 2000; Nadjar, 2000; Pagani et al., 2000; Schønheyder et al., 2000; Tzouvelekis et al., 2000; Mathers et al., 2015; Popescu et al., 2017). This repertoire of MDR then necessitates the use of carbapenams as the only useful chemotherapeutic option to treat ESBL-linked infections. However, since the 2000s, an unprecedented crisis involving carbapenemases spreading globally and including genotypes such as bla NDM, bla KPC, bla IMP, bla OXA, and bla VIM has been observed and reported upon (Liu et al. 2009; Curiao et al. 2010; Cuzon et al. 2010; Pournaras et al. 2010; Prior et al. 2010; Lee and Burgess, 2012; Tzouvelekis et al. 2012; Chen et al. 2014b; Hudson et al. 2014; Jin et al., 2015; Mathers et al. 2015; Zhang et al., 2015; Zheng et al., 2016). Those plasmids associated with carbapenem resistance-encoding genes often carry other resistance- and virulence-encoding mechanisms (Huang et al. 2013; Hudson et al. 2014; Hennequin and Robin, 2016; Zheng et al., 2016). This situation has been further complicated with the identification of the transmissible colistin resistance genes mcr-1 (Liu et al., 2016), mcr-2 (Xavier et al., 2016), and mcr-3 (Yin et al., 2017), together with chromosomal mutations in mgrB, pmrA, pmrB, phoP, and phoQ (Choi et al., 2016). Increasing numbers of reports are describing the mcr-1 gene in K. pneumoniae of human origin (Gu et al., 2016; Leangapichart et al., 2016; Rolai et al., 2016; Stoesser et al., 2016; Newtonfoot et al., 2017; Quan et al., 2017; Tian et al., 2017; Mendes et al., 2018), environmental origin (Zhao et al., 2017), fruit origin (Yang et al., 2019), and (food producing) animal origin (Davis and Price, 2016; Tian et al., 2017; Wu et al., 2016; Argudín et al., 2017; Kieffer et al., 2017; Saidani et al., 2019). Furthermore, KPCs producing K. pneumoniae have already been detected and which co-expressed colistin resistance carrying mcr-1 (Li et al., 2016; Aires et al., 2017; Novović et al., 2017). This finding highlights the potential for dissemination of KPC-producing K. pneumoniae carrying an mcr-1 gene and this development may be associated with MDR international clones. Continued selection for the transmission of these genes could pose a threat to infection control strategies and future clinical treatments.
Polymyxins and tigecycline are regarded as major treatment options for carbapenem-resistant Enterobacteriaceae (CRE) infections (Sheu et al., 2019). However, the emergence and global dissemination of mobile colistin resistance has limited the efficacy of polymyxins (Liu et al., 2016), and tigecycline consequently is one of the few remaining drugs of choice in cases of severe CRE infection. Tigecycline is an important compound used in the treatment of CRE infection, especially carbapenem-resistant K. pneumonia (CRKP) (Li et al., 2015). However, continued clinical use of tigecycline led to a reduction in its efficacy, which in turn is linked to high mortality rates resulting from CRKP, the most common type of CRE (Navon-Venezia et al., 2017; Wang et al., 2018; Lv et al., 2020). More recently, a new plasmidborne RND efflux pump-type tigecycline resistance determinant was identified and found to be widespread among K. pneumoniae isolates from food-producing animal origin, offering the potential for onward transmission (Lv et al., 2020).
Most of the large plasmids identified in K. pneumoniae have been found to be associated with increased morbidity and mortality (Ramirez et al., 2014). It has been suggested that K. pneumoniae cultured from blood isolates carry a 200-kbp plasmid, which includes iron transport systems and the mucoid phenotype-encoding genes (Chen et al., 2004). Recently, two plasmid-driven epidemics in which the genes bla KPC and bla NDM were combined in the same bacterium have been reported (Pesesky et al., 2015; Quiles et al., 2015; Zhu et al., 2015), creating superbugs that are refractory to chemotherapeutic management.
It is thought that the acquisition of multiple plasmids from MDR isolates could be associated with limited fitness cost (Sheppard et al., 2016; Buckner et al., 2018; Wyres et al., 2019; Nang et al., 2018). Interestingly, data showed that MDR K. pneumoniae clones could more easily acquire virulence genes than hypervirulent clones could acquire resistance genes (Wyres et al., 2019), indicating a greater need for global genomic surveillance encompassing AMR information of K. pneumoniae.
Treatment of K. pneumoniae Infection
AMR in MDR K. pneumoniae is an important public health issue for patients in clinical setting, thereby limiting optimal treatment options (Bassetti et al., 2018). More treatment failures are recorded in those cases receiving monotherapy, when compared to others who received combination therapy (49% vs. 25%; p = 0.01) (Lee et al., 2012). In terms of the higher level of antimicrobial resistance in Klebsiella and the increasing incidence of CRKP, combination therapies using carbapenems, polymyxins, tetracyclines, and fluoroquinolones are recommended and widely used. There were no major treatment effect differences recorded between the three most common antibiotic class combinations: polymyxin plus carbapenem, polymyxin plus tigecycline, and polymyxin plus aminoglycoside (Lee et al., 2012; Petrosillo et al., 2013; Bassetti et al., 2018). Nevertheless, the best combination for each infection type remains to be confirmed and the continued use of carbapenems in combination therapy is likely to continue (Petrosillo et al., 2013).
In situ Identification of Foodborne K. pneumoniae and Rapid Detection of MDR Phenotypes by Raman Spectroscopy
Conventional approaches for testing drug resistance in bacteria usually requires the monitoring of bacterial growth during incubation with different antimicrobial compounds. Although the methodology is standardized, this is time-consuming, delaying the diagnosis and subsequent prescription of an alternative and more efficient drug if indicated. Whole-genome sequencing techniques and deep-level transcriptomics are powerful tools to identify, localize, and explore the expression of MDR genes in pathogens using high-throughput sequencing platforms. These developments now make it feasible to screen specific genotypes in a given bacterial isolate. Analysis of raw sequencing data, however, still requires a mastery of several appropriate software programs. Moreover, in the context of rapidly evolving bacterial populations, and for K. pneumoniae in particular, repeated exposure to a large range of antimicrobial compounds can trigger the emergence of new MDR phenotypes, for which the susceptibility to the available antimicrobial drugs remains unknown.
To choose the most efficient drug for treatment, screening techniques are needed to rapidly test the efficacy of a set of antimicrobial compounds against a bacterial isolate of interest, as quickly as possible. This test, if possible, would provide information on the drug susceptibility of the bacterium without prior characterization of the species, and on a limited number of cells to reduce the duration of the growth phase before the analysis. Raman spectroscopy could potentially fill this gap.
Several reviews highlighted the power of Raman spectroscopy for the identification of bacteria (Pahlow et al. 2015; Stöckel et al. 2016; Lorenz et al. 2017), which is a label-free, nondestructive vibrational spectroscopy technique that can be combined with confocal microscopy for imaging biological samples. The Raman spectrum of a single live bacterium can be acquired within a few minutes with limited sample preparation. This analysis unveils the molecular fingerprint of the bacterium, providing access to broad information content describing the chemical composition and the structure of the biomolecules (DNA/RNA, proteins, lipids, and carbohydrates) within the sample. The spectral signature of one bacterium is the result of the unique combination of molecular vibrations of these biomolecules and can be used to identify groups and subgroups of bacteria (Stöckel et al. 2016). Liu et al. (2009) showed that K. pneumoniae exhibits a different Raman spectrum compared to Enterococcus feacalis, Listeria monocytogenes, Staphylococcus aureus, Mycobacterium tuberculosis, Mycobacterium gordonae, Escherichia coli, and Serratia marcescens. The identification and subtyping of bacteria in complex matrices or in line during food processing remain challenging, Raman spectroscopy represents a powerful tool for the detection of foodborne pathogens in situ (Witkowska et al. 2017; Yaseen et al. 2017; Zhao et al. 2018).
Raman spectroscopy has indeed become an easy-to-use, high-speed analysis technique for the identification of pathogens with applications in medicine, and more laterally in food safety (Yaseen et al. 2017). The main advantages are (i) instantaneous analysis, (ii) limited sample preparation, (iii) in situ analysis in complex matrices, and (iv) real-time analysis of live bacteria.
When combined with powerful multivariate statistical and discriminant analysis, Raman spectroscopy also allows discrimination of pathogenic and nonpathogenic bacterial subtypes (Hamasha et al. 2013). A Raman study list of β-lactamases has been summarized, which included SHV1/2/5, OXA1/24 β-lactamases, and carbapenemases, among others (Carey et al. 2015). Clones expressing different resistance phenotypes against antimicrobial compounds (cephalosporins and quinolones) could also be successfully identified among K. pneumoniae (Guyot et al. 2012; Cheong et al. 2017). Using the highly specific spectral signature of each bacterium, Raman imaging could further be used to screen for MDR bacteria embedded in a food matrix. This approach has great potential in food safety and quality control, opening the way to the on-line detection and identification of largely overlooked foodborne bacteria, provided that robust data analysis routines are implemented.
A variant of Raman spectroscopy called Surface-Enhanced Raman Scattering (or SERS) has proven very efficient in probing the cell wall changes of bacteria due to its capacity to locally enhance the Raman signal of the bacterial cell wall components. Therefore, discrimination between Gram-positive and Gram-negative bacteria is reproducible and any disruption of the cell wall or cell membrane can be assessed by real-time analysis of the live bacteria after incubation with different antimicrobial compounds (Prucek et al. 2012; Liu et al. 2009; Hlaing et al. 2018). Results have also been obtained by conventional Raman spectroscopy (Xuan Nguyen et al. 2017). As several antimicrobial compounds specifically target the bacterial cell membrane, this approach is promising for the rapid screening of the susceptibility of a bacterial isolate to a large set of antimicrobial drugs in a clinical or food safety context (Cheong et al. 2017). The mechanistic investigation of the effect of antimicrobial drugs by Raman spectroscopy could benefit in the future from the development of other techniques based on the Raman effect, such as SERS, Coherent Anti-Stokes Raman Scattering, and Stimulated Raman Scattering (SRS) (Evans and Xie, 2008; Hong et al. 2018).
When transmission occurs throughout a hospital, there is an urgent need for the use of a rapid subtyping strategy designed to detect and subsequently prevent further pathogen spread. Voor in't Holt et al. (2015) reported a clonal outbreak of ESBL-producing Klebsiella in a hospital. They concluded that subtyping should be continuously performed not only in the case of outbreaks but also in an apparent non-outbreak situation. Raman spectroscopy could be the rapid typing method easy to implement as an early identification and an immediate infection-prevention strategy to prevent further transmission of those small outbreaks identified early on.
Virulence and Pathogenicity in K. pneumoniae
As mentioned above, the GI tract is an important site for colonization of Klebsiella species (Fung et al., 2012). It is predicted that the majority of Klebsiella infections follow a similar pattern in humans as demonstrated by other food-poisoning pathogens, such as E. coli, L. monocytogenes, Salmonella species, or Campylobacter species (Strauman et al., 2010; Robinson et al., 2012; Gessain et al., 2015; Kirk et al., 2016).
K. pneumoniae employs various virulence factors (VFs) and strategies to survive in the host infection process. Paczosa and Mecsas (2016) reviewed four major classes of VF, which were well studied and described in K. pneumoniae, including capsule, lipopolysaccharide (LPS), siderophores, and fimbriae. Liu et al. (2019) separated and resolved the VFs of K.pneumoniae into several further detailed groups, especially for fimbriae and siderophores, with supplementary VF types in the virulence factor database (
Virulence Factors and Related Virulent Genes in Klebsiella pneumoniae (Modified and Updated Based on Virulence Factors in Virulence Factor Database)
LPS, lipopolysaccharide.
There are several ways by which Klebsiella species can be transmitted to susceptible individuals; infections can be transmitted through contaminated food or water, particularly in CA-type scenarios (Davis and Price, 2016). The bacterium can also be transmitted through animal-to-person contact, person-to-person contact, or air droplets (Shon et al., 2013). The latter two routes are the most common means involved in HA cases of infection (Ridolfo et al., 2016).
Extraintestinal infections are a common event caused by Klebsiella and, in particular, linked with hypervirulent isolates. These bacteria are able to ascend into the bladder from the perineum (Dedeić-Ljubović and Hukić, 2009), or translocate the GI tract into the peritoneal cavity or other extraintestinal locations to cause PLA among other metastatic infections, such as meningitis and endophthalmitis (Liu et al. 2013; Hsu et al., 2015; Kazanji et al., 2016). It is reported that translocation through the GI tract could be the most likely route leading to K. pneumoniae causing PLA (Chung et al., 2011; Fung et al. 2012). Hypervirulent K. pneumoniae (hvKp) is now becoming increasingly recognized as a pathogen of concern. Some of the distinguishing phenotypes may include emergence of XDR strains, an increase in capsule, and siderophore production by hvKp relative to classical K. pneumoniae (Sellick and Russo, 2018). However, specific studies describing the epidemiology and transmission of hvKp are lacking.
These following lines will focus on the manner in which Klebsiella causes extraintestinal infections such as PLA, or bacteraemia through the oral route by crossing the intestinal mucosa layer.
Infection Pathway Traversed by Klebsiella species
When food contaminated with Klebsiella is ingested, the pathogen will transit through the GI tract to colonize the intestine (Fig. 1A). Along this pathway, Klebsiella will be exposed to several hydrolytic enzymes, variations in pH, and competition with other bacteria together with the host's immune system. K. pneumoniae, like other pathogens, is susceptible to the action of phospholipases, amidases, and especially lysozyme, a common enzyme present in the mucosal surfaces that degrades peptidoglycan (Markart et al., 2004). The presence of stomach and bile acids presents an additional barrier to bacterial survival. Bile acids are released into the duodenum and are found along the GI tract (Anes et al., 2015). The goblet cells of the epithelial barrier are responsible for the production of highly glycosylated mucins and other mucin-derived products that are the most abundant proteins in the intestinal lumen (Fig. 1B).
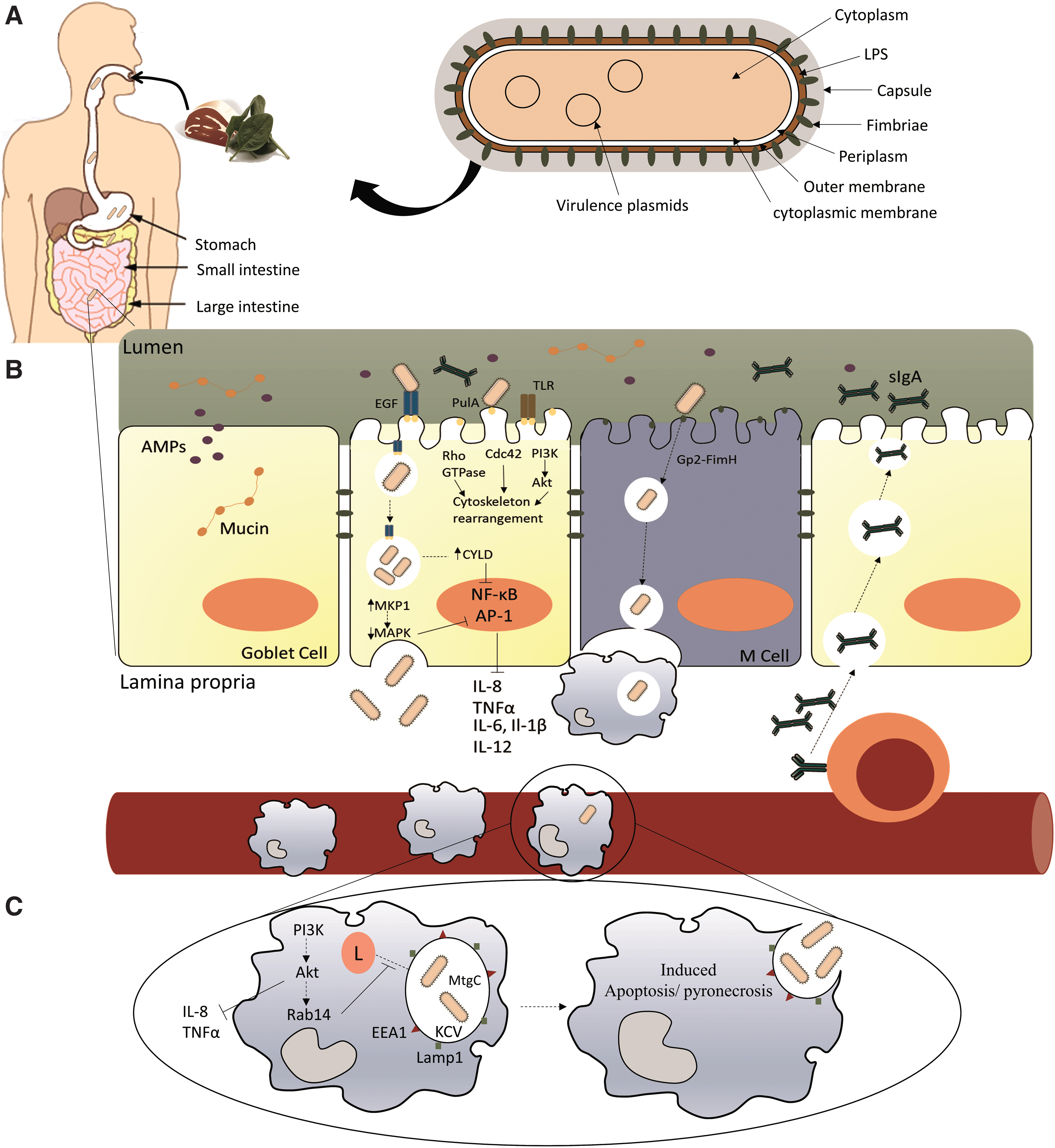
Antimicrobial proteins (AMPs), such as defensins, cathelicidins, and lysozyme, are released into the lumen mucus by Paneth cells. Together with the presence of immunoglobulin A, they constitute an initial line of defence against pathogens (Peterson and Artis, 2014). Gram-negative bacteria also possess intrinsic efflux mechanisms such as AcrAB-tolC, which aid their survival in harsh environments (Anes et al., 2015). In Klebsiella species, Klebsiella-specific efflux pumps such as EefXABC (Coudeyras et al., 2008), which confers resistance to inorganic acids and hyperosmolarity (the same acidic conditions that can be found in the stomach), or the KpnEF (Srinivasan and Rajamohan, 2013) and KpnGH (Srinivasan et al., 2014) pumps are also active and these can help the pathogen survive in the presence of AMPs and bile stress, giving the bacterium time to adapt to the environment. To overcome these host defence mechanisms, particularly the enzymatic-mediated strategies, Klebsiella undergoes several modifications of its capsule, peptidoglycan, and LPS so the hydrolytic activity of these enzymes is compromised (Moranta et al., 2010; Hsieh et al., 2013; Bachman et al., 2015; Llobet et al., 2015). It has also been reported that the genes pgaABCD, which are involved in the synthesis and secretion of poly-β-linked N-acetylglucosamine, mediate biofilm formation under bile salt stress (Chen et al. 2014a). The stress induced by all these defence mechanisms works also as a signaling trigger to prepare the bacteria for the next step, the colonization of the epithelial cells. Urea is found in the gut in abundance; the enzymatic ability to hydrolyze urea to ammonia is due to the presence of the ureDABCEFG operon, which contributes to the active growth of K. pneumoniae, improving its ability to colonize the host's GI tract (Maroncle et al., 2006). Another metabolic advantage expressed by Klebsiella is the ability to metabolize allantoin to carbon and nitrogen. The allantoin operon has been found to be associated with hypervirulent strains causing liver abscess (Yu et al., 1991; Chou et al., 2004).
Adhesins and biofilm formation in K. pneumoniae
To colonize the intestine, Klebsiella requires the necessary machinery to adhere to epithelial cells. Adhesins play an important role in initiating the process of colonization; however, not all are involved in adhering to the host's cells directly. There are six types of fimbriae in Klebsiella responsible for this process: type 1 fimbriae, type 3 fimbriae, Kpc fimbriae, E. coli common pilus (ECP), KPF-28 adhesin, and the nonfimbrial antigen CF-29K.
Type 1 fimbriae are encoded by the locus fimBEAICDFGHK and are common in all Enterobacteriaceae. These structures are thin, thread-like surface protrusions located on the outer membrane, composed of repeating FimA subunits with a FimH molecule at the base and whose expression is regulated by fimK (Struve et al., 2009). Other regulators such as bamB appear to be also involved (Hsieh et al., 2016). The remaining genes within this cluster, fimF and fimG, code for small structural subunits that confer some variability to these fimbriae. The genes fimB and fimE encode two recombinases that are responsible for the switch between the fimbriated and nonfimbriated states. Similarly, fimC encodes for a fimbrial chaperone and fimD is expressed as an usher protein (Struve et al., 2008; Paczosa and Mecsas, 2016). These fimbriae bind to D-mannosylated glycoproteins present in the host or extracellular matrix surfaces (Stahlhut et al., 2009).
Type 3 fimbriae are encoded by the mrkABCDFJIH gene cluster and it is present in almost all K. penumoniae (Langstraat et al., 2001). In a similar arrangement as for the structure of type 1 fimbriae, type 3 fimbriae consist of several MrkA subunits protruding from the outer membrane in a helix-like structure, with the adhesion MrkD located at the tip. MrkB is a chaperone and mrkC encodes an usher protein (Struve et al., 2009), while MrkF is involved in the surface stability of the fimbriae. The remaining genes mrkJIH are involved in regulation and expression under different conditions (Johnson et al., 2011; Tan et al., 2015). Type 3 fimbriae are mannose resistant in contrast to type 1 fimbriae. These structures mediate attachment in vitro to human endothelial and urinary bladder cell lines (Tarkkanen et al., 1997), and in coated matrices with proteins such as type IV and type V collagen (Tarkkanen et al., 1990; Jagnow and Clegg, 2003).
Notably, type 1 and type 3 fimbriae have no effect on the ability of K. pneumoniae to colonize the intestine or the lung. Type 1 fimbrial genes were found to be upregulated in urinary tract infections (Struve et al., 2008, 2009), a similar observation also being made for type 3 fimbriae that have the ability to colonize the bladder (Tarkkanen et al., 1997; Murphy et al., 2013). Fimbriae of type 3 also seem to play an important role in biofilm formation (Schroll et al., 2010). Biofilm formation is an important step in protecting bacteria from the epithelial antimicrobial agents, conferring an improvement in the ability of Klebsiella to survive in the intestine. The ability of K. pneumoniae to bind to surfaces such as indwelling catheters or other medical devices provides it with an ability to seed vulnerable sites and to persist in patients (Paczosa and Mecsas, 2016). Deletions of both fimbriae type 1 and type 3 do not appear to attenuate virulence in a lung infection model (Struve et al., 2009), indicating that other factors and/or fimbriae play a role in infection.
Another fimbria that confers strong biofilm forming activity is the Kpc fimbriae. This type of fimbriae is encoded by the kpcISABCDEFG operon and act in a similar manner as the type 1 and type 3 fimbria systems (Wu et al., 2010). Interestingly, this system was not found to be expressed under any in vitro condition studied, leading to speculation about a specific colonization condition or environmental stress. Kpc fimbriae were found to be most prevalent in clinical isolates expressing hypervirulent phenotypes (Wu et al., 2010).
The ECP homolog, present in the K. pneumoniae genome, is characterized by the operon ecpABCDE and has been shown to be involved in cell adherence, maintaining a strong physical contact between the host and the bacteria (Alcántar-Curiel et al., 2013). Currently, little is known about this pili system in K. pneumoniae.
KPF-28 adhesin is a long, thin flexible fimbria located on a transmissible R plasmid that also encodes the CAZ-5/SHV-4 β-lactamase enzyme, and the former facilitates adherence to Caco-2 cells in vitro. The first time these fimbriae were reported was in Claremont-Ferrand, France, in clinical urinary tract infection (UTI) cases (Di Martino et al., 1996). Another example of a nonfimbrial adhesin capable of adhering to Caco-2 cells is the CF-29K. The gene encoding this 29-kDa protein was located on a conjugative plasmid in K. pneumoniae (Darfeuille-Michaud et al., 1992; Di Martino et al., 1995). Little is known about the manner by which these fimbriae and non-fimbriae function to attach the bacterium to intestinal cells. It has been reported that the Klebsiella capsule can impede cell adherence and invasion, whereas LPS contributed to adhesion in a nonspecific manner (Sahly et al., 2000; Clements et al., 2008).
Although the correlation between fimbrial expression and the attachment of the pathogen to the host's intestine is not fully elucidated, it is clear that the formation of biofilm renders K. pneumoniae more resistant to the host defences and improves its capability to colonize epithelial cells. The formation of a biofilm, in addition to the presence of fimbriae described above, involves metabolic and structural changes in the capsule and LPS (Balestrino et al., 2008). K. pneumoniae biofilms are mainly composed of mannose (40–50%) (Bales et al., 2013). In vitro studies indicate that genes involved in amino acid synthesis and in
The role of the capsule (CPS) in Klebsiella species
The initial downregulation of capsule gene expression is necessary for the bacteria to attach the host cells (Clements et al., 2008). The Klebsiella capsule is considered one of the major virulence effectors. The CPS cluster is composed of many genes involved in its synthesis (wzi, wza, wzb, wzc, gnd, wca, cpsB, cpsG, and galF) and these are collectively known as the K-antigens (Paczosa and Mecsas, 2016). From the 77 described capsular (K) types, those of K1, K2, K4, and K5 are the most important and are associated with virulent isolates (Brisse et al., 2009). Capsule production can also be enhanced by the presence of transcriptional regulators rmpA and rmpA2 found on MGEs, among the common regulators of the CPS cluster (rcsA and rcsB) (Paczosa and Mecsas, 2016). Two other important genes involved in regulation include rfaH that encodes a transcriptional elongation factor and which was found to be important in a murine lung infection model (Bachman et al., 2015), and the htrA gene that codes for a serine protease involved in the capsule formation (Cortés et al., 2002). The complete mechanism by which CPS is regulated remains to be further elucidated.
Infection of epithelial cells
To infect internal organs, Klebsiella must cross the epithelial barrier (Fig. 1B). There are two different routes used to enter epithelial cells: the intracellular route in which the pathogen invades nonphagocytic epithelial cells, altering the host cytoskeleton (Cossart and Sansonetti, 2004); and the paracellular pathway, which involves the disruption of the tight junctions, allowing the pathogen to pass between epithelial cells (Okuda et al., 2010; Backert et al., 2013). M cells are also considered to be an entry route to cross the epithelial barrier. These special cells are responsible for presenting foreign antigens and intact bacterial cells to the macrophages and dendritic cells (Peterson and Artis, 2014). When the glycoprotein GP2 present on the surface of the cells binds to antigens or FimH, as it happens with E. coli or Salmonella, it activates transport across the epithelial barrier for presentation to the phagocytic cells (Hase et al., 2009).
K. pneumoniae has been shown to invade and replicate inside a nonphagocytic epithelial cell in an in vivo model and to translocate in vitro across the epithelial monolayer without damaging the integrity or permeability of the barrier (Hsu et al., 2015). The mechanism by which Klebsiella initiates the invasion is still unresolved; however, data suggest that Klebsiella interacts with the glycans in the epithelial cell through the type II secretion system referred to as PulA (Tomás et al., 2015). Through this mechanism, Klebsiella is able to reduce the inflammatory response by avoiding the TLR2–TLR4–MyD88 pathway. Toll-like receptors (TLR) are the common mechanism for bacterial recognition in epithelial cells. Normally, when activated, this pathway will trigger the activation of the nuclear factor-κB (NF-κB), which induces the activation and release of cytokines (Moranta et al., 2010; Peterson and Artis, 2014). PulA interaction with the epithelial cell causes the activation of the Rho GTPase, Cdc42, and the EGF–PI3K–Akt pathway, inducing cellular cytoskeleton rearrangements, thereby allowing the bacterium to translocate (Frank et al., 2013; Hsu et al., 2015). This mechanism is also used by Salmonella species and Shigella species, wherein a type III secretion system deploys effector proteins into the host cell that will cause structural rearrangements, thereby facilitating the microorganism to be internalized (Cossart and Sansonetti, 2004). Klebsiella polysaccharides play an important role during the infection mechanism. Apart from antimicrobial defence, CPS also plays a part in interfering with the TLRs on the cell surface during infection (Moranta et al., 2010). It appears that CPS might have a dual role by activating the EGF–PI3K–Akt and inhibiting the Rac1 pathway (Regueiro et al., 2011; Frank et al., 2013). This inhibition causes an increased expression of NOD1, which in turn upregulates the expression of CYLD and MKP-1, allowing Klebsiella to highjack two host systems used by the epithelial cells to return to homeostasis, the NF-κB and the MAPKs pathways, respectively (Regueiro et al., 2011). The LPS O-polysaccharide and the first glucose of the LPS core also aid in dampening down the inflammatory response of the epithelial cell by disturbing TLR recognition of Klebsiella, attenuating the activation of NF-κB (Tomás et al., 2015). Klebsiella is able to alter its lipid A composition in the presence of different tissues in a PhoPQ-regulated, LpxO-dependent manner (Llobet et al., 2015) and by ramA activation (De Majumdar et al., 2015).
The iron acquisition system in Klebsiella species
When inside the epithelial cell and in plasma, Klebsiella encounters an extreme environment in terms of iron availability. Iron is an essential co-factor for bacterial growth and replication and it has been implicated in virulence (Bialek-Davenet et al., 2014). One of the host's innate mechanisms when infected is to sequester the readily available iron to restrict the growth of the pathogen. In plasma, transferrin is responsible for transporting iron, thereby limiting its availability. In the case of bacterial infection, this system can be improved by shifting the binding of iron to lactoferrin (Bullen et al., 1972). To cope with the low levels of iron, Klebsiella is able to synthesize and secrete small iron-scavenging molecules known as siderophores, which acquire and deliver host iron to the bacterium, resulting in bacterial growth. In addition, siderophores also induce cellular stress responses related to the stabilization of the master transcription factor hypoxia-inducible factor-1α, ultimately resulting in bacterial dissemination to the spleen (Holden et al., 2016). Together with the two ABC efflux systems, Kfu (Lee et al., 2016) and Sit (Sun et al., 2014), and the receptor tonB (Hsieh et al., 2008), these iron-scavenging mechanisms are required for full virulence (Russo et al., 2014; Holt et al., 2015).
Several siderophores are produced by Klebsiella species, including enterobactin, yersiniabactin, salmochelin, and aerobactin, and some of these are encoded on the chromosome, whereas others are commonly found in MGEs (Paczosa and Mecsas, 2016).
Enterobactin is the siderophore with highest affinity for free iron and is encoded on the chromosome by the genes entABCDEF (biosynthesis system) and fepABCDG (transport system) (Crichton, 2016). The action of this siderophore can be neutralized by lipocalin-2 (Bachman et al., 2012). To counteract the action of the host, Klebsiella and other Gram-negative bacteria incorporate other siderophores to scavenge iron from the host. Yersiniabactin was initially identified in Yersinia species as part of the high pathogenic island (Carniel, 1999). This siderophore is encoded by the irp cluster and is associated with invasive Klebsiella infections, being one of the most prevalent virulence loci identified in hvKP (Carniel, 2001; Bachman et al., 2011; Holt et al., 2015). Although yersiniabactin is not affected by the action of lipocalin-2, this siderophore is unable to acquire iron in the presence of transferrin (Bachman et al., 2011).
Like yersiniabactin, aerobactin and salmochelin are also examples of an alternative system that is nonresponsive to the action of lipocalin-2. Aerobactin is a citrate-hydroxamate siderophore encoded by the genes iucABCD along with the cognate transporter iutA (Bailey et al., 2016) and salmochelin, an O-glycosylated form of enterobactin, encoded by the genes iroBCDE and the transporter iroN (Müller et al., 2009). Both are associated with hypervirulent Klebsiella infections (Hsieh et al., 2008; Russo et al., 2014; Holt et al., 2015) due to the presence of these clusters on virulence plasmids, which are often co-located with resistance markers such as the rmpA and kfuABC or the two-component regulatory system kvgAS (Nassif and Sansonetti, 1986; Darfeuille-Michaud et al., 1992; Chen et al., 2004; Wu et al., 2009; Russo et al., 2015). The presence of such siderophores increases the infection potential of K. pneumoniae.
Resistance/evasion to host immune system
Human plasma carries many proteins capable of binding to pathogen-expressed antigens and that subsequently activate the Complement system. The Complement system is also part of the innate defence mechanism of the host. The LPS and outer membrane proteins (OMP) are the primary targets (Albertí et al., 1993, 1996). Klebsiella CPS once more plays an important role as a barrier preventing the binding of antibodies or the C1q by masking the epitopes present in the bacterial cell wall, thereby blocking its activation (de Astorza et al., 2004; Lin et al., 2012; Lee et al., 2014).
OMPs are the main target of the membrane attack complex (MAC). LPS plays an important role in stabilizing the OMPs by protecting them from the external environment (Needham and Trent, 2013). Modifications in the LPS structure have been shown to cause changes in serum resistance in Klebsiella (Williams et al., 1983; Ciurana and Tomás, 1987). The same susceptibility to serum was detected when the transcriptional elongation factor rfaH, responsible for LPS regulation, was deleted (Bachman et al., 2015). Klebsiella can undergo several modifications in the LPS to evade host immune detection (Llobet et al., 2015). The elongation of the O-LPS leads to the deposition of C3b further away from the cell wall, preventing the proper localization of the MAC on the bacterial surface, resulting in reduced pore formation (Merino et al., 1992).
Another strategy used by Klebsiella is the production and secretion of LPS-containing outer membrane vesicles into the environment to absorb proteins of the Complement system, preventing the subsequent deposition of these proteins onto the bacteria surface (Lee et al., 2012; Turner et al., 2016).
Although all these mechanisms help bacteria to avoid macrophage detection inevitably, macrophage engulfment will take place either by presentation of antigens by M cells or by direct capture in the plasma. Nonetheless, Klebsiella seems to be adapted to survive macrophage hydrolytic activity, a feature also found in other pathogens (McClure and Schiller, 1996; Ruckdeschel et al., 1997; Richardson, 2015; Srikumar et al., 2015).
It has been suggested that Klebsiella might trigger the lipid rafts to favor internalization by the macrophage (Huang et al. 2013). Klebsiella is able to survive inside the macrophage in the Klebsiella-containing vacuole (KCV) by deviating from the canonical endocytic pathway through manipulation of the PI3K–Akt–Rab14 (Fig. 1C). Interestingly, the KCV was able to acquire the markers of the endocytic pathway EEA1 and Lamp1. By manipulating this pathway, Klebsiella can control phagosome maturation by blocking the fusion of the KCV with the lysosome (Cano et al., 2015). The acidic pH of the KCV triggers the downregulation of the CPS, possibly due to the high metabolic burden to maintain such a system, proving that the presence of the capsule is not necessary for survival inside the macrophage (Cano et al., 2015). The VF MgtC used by Salmonella to inhibit the activity of the bacterial F1F0 ATP synthase, thereby disrupting proton translocation, allows the bacterium to adapt to the acidification of the vacuole and restores metabolic imbalances (Lee et al., 2013). The Klebsiella MgtC protein is very similar to a homolog in Salmonella; so it is speculated that this protein might have the same function (Belon and Blanc-Potard, 2016). Klebsiella is also able to induce programmed macrophage death. Nevertheless, the mechanism by which it happens is not fully elucidated. While some authors report features of cell death similar to apoptosis (Cano et al., 2015), others reported features of pyronecrosis by activation of NLRP3-dependent pathway (Willingham et al., 2009; Webster et al., 2010). One possible reason for such variability could be the different experimental conditions used. Further studies are required to clarify the cell death mechanism used by K. pneumoniae.
Conclusions
As a hitherto incompletely recognized zoonotic bacterium, K. pneumoniae is responsible for a wide range of human CA and HA infections, involving not only humans, animals, and environments but also the food chain. MDR and hypervirulent KpI (hvKP) strains have been increasingly evolving into distinct CGs, demonstrating a broad capacity to infect host cells, and rendering infection by these strains, to be very challenging to treat. MGEs carrying MDR or virulence determinants contribute to the potential for rapid dissemination of K. pneumoniae. Continued studies on K. pneumoniae deciphering its mechanism of resistance, pathogenicity, physiology, and interactions with host tissues should provide insights into useful strategies to combat this pathogen of importance to human health.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.