
Abstract
This study reports a genomic analysis of Escherichia coli isolates recovered from 25 bovine fecal composite samples collected from four different production units in Maputo city and around Maputo Province, Mozambique. The genomes were analyzed to determine the presence of antibiotic resistance genes (ARGs), genetic relatedness, and virulence factors known to cause diseases in humans. Whole-genome sequencing was conducted on 28 isolates using an Illumina NextSeq 500 sequencing platform. The genomes were analyzed using BLASTN for the presence of resistance genes and virulence factors, as well as to determine their phylogenetic groups, sequence types (ST), and ST complexes (ST Cplxs). The majority of the isolates (85%) were identified as members of phylogenetic groups B1, with fewer isolates identified as members of group A, and a single isolate identified as group “E/Clade I.” The ST analysis demonstrated a higher level of diversity than the phylogenetic group analysis. Sixteen different STs, five ST Cplxs, and seven singleton complexes were identified. A strain identified as a novel ST (ST9215) showed a high level of similarity with an isolate recovered from a wild animal in the Gambia. Seven different ARGs were identified, with tet(B) being the most frequently detected, followed by aph(3″)-Ib, aph(6)-Id, sul2, bla TEM-1B, and dfrA1. Three isolates encoded β-lactam-conferring point mutations in the ampC promoter (−42C>T). In total, 51 different virulence factors were identified among the genomes. This study demonstrates that E. coli from bovine sources in Mozambique encoded multiple antibiotic resistance elements, plasmids, and virulence factors. To the best of our knowledge, this is the first genomic description of antibiotic-resistant E. coli isolated from bovine sources in Mozambique.
Introduction
E
Antimicrobial resistance (AMR) is often observed in both pathogenic and nonpathogenic E. coli, including commensal strains isolated from animals (Bosman et al., 2012; Schmidt et al., 2015; Cao et al., 2019, Salaheen et al., 2019; Springer et al., 2019; Oh et al., 2020). Resistant pathogenic strains may cause intractable infections in humans and animals, and resistant nonpathogenic strains may cause opportunistic infections or can potentially transfer resistance elements to pathogenic Enterobacteriaceae (Oladeinde et al., 2019). AMR remains a serious human and animal health issue on a global scale causing an extremely high, but not yet quantified, number of deaths per year (ECDC, 2017; CDC, 2019; O'Neill, 2014). To address the AMR burden, a framework has been developed in some countries for the judicious veterinary use of antibiotics that are medically important for humans (FDA, 2015). However, resistant bacteria from human and animal sources have not been fully described in many countries, thus leaving significant gaps in the knowledge of the true global burden of AMR.
In recent decades, low- and middle-income countries experienced an increase in selective pressure for the emergence of AMR, likely due to the lack of regulation of antimicrobial agents, prescription practices outside clinical settings, availability of antimicrobials on the informal market, and campaigns administering antimicrobials to healthy people (Berendes et al., 2019). In Mozambique, the burden for treatable communicable diseases remains high for humans and animals. In a country where half of the population lacks access to a primary health care facility, often the informal sector represents the only feasible point of access to antimicrobials for both humans and animals. The use of nonprescribed antibiotics, inappropriate use, and inconsistent knowledge about antibiotics and antibiotic resistance is an increased concern for the spread of AMR in this country (Berenedes et al., 2019; Mate et al., 2019; Torres et al., 2019; Cambaco et al., 2020; Rodrigues, 2020). Even though limited information on resistance rates is available, recent studies conducted on human infections have documented resistance to first-line antibiotics in many pathogens (Mandomando et al., 2009, Mandomando et al., 2010, Ibarz-Pavón et al., 2011, Mashana et al., 2013, Berendes et al., 2019) and in bacteria isolated from the environment (Taviani et al., 2008). Furthermore, the presence of AMR in animal agriculture, where antimicrobials are used for disease prevention and growth promotion, is insufficiently documented. A genomic evaluation of resistant bacteria in bovine feces has not been described at the genomic level in Mozambique. To investigate the genomic characteristics of E. coli isolates recovered from bovine sources around Maputo, Mozambique, we sequenced the genomes of a subset of resistant and susceptible isolates collected during a larger concurrent study.
Materials and Methods
Composite manure was collected between April and May 2017 from a cross-sectional sampling of four locations, identified as Farms 1 to 4, in and around Maputo, Mozambique (population >1.3 million people). The four farms included in the study were small units that raised animals for both beef and dairy production. Animal management protocols, including antibiotic usage, were not made available. At the time of sample collection, the number of animals on Farms 1 to 4 was 34, 4, 87, and 23, respectively. At each farm, ∼120 g of manure was collected from at least five sites within the pen and combined into a single composite sample (∼750 g) from which an aliquot was suspended in buffered peptone water (5 g in 45 mL) and streaked onto CHROMagar E. coli (CHROMagar, Paris, France). At least five randomly selected, presumptive E. coli colonies from each sample were transferred to Simmons Citrate Agar (BD Diagnostics), MacConkey Agar (Remel, Lenexa, KS), and L-Agar plates and incubated at 37°C for 24 h. Sensitivity to 14 antibiotics on the National Antimicrobial Resistance Monitoring System for Enteric Bacteria (NARMS) GN panel (CMV3AGNF) (Table 1) was determined using an automated antimicrobial susceptibility system (Trek Diagnostic Systems, Westlake, OH). Cutoff values for antibiotic susceptibility were determined using the Clinical and Laboratory Standards Institute-established breakpoints or the NARMS reference values (CDC, 2018; Cassini et al., 2019; CLSI, 2019). Reference strains for the antibiotic sensitivity analyses were Enterococcus faecalis ATCC 29212, Staphylococcus aureus ATCC 29213, E. coli ATCC 25922, and Pseudomonas aeruginosa ATCC 27853.
Phenotypic Antibiotic Susceptibility Data for Isolates Selected for Genome Sequencing
Minimum inhibitory concentrations are expressed in μg/mL. White square represents susceptible, and black square represents resistant.
Breakpoint (μg/mL).
AMP, ampicillin; ARS-CC; AUG, amoxicillin/clavulanic acid; AXO, ceftriaxone; AZI, azithromycin; CHL, chloramphenicol; CIP, ciprofloxacin; FIS, sulfisoxazole; FOX, cefoxitin; GEN, gentamicin; NAL, nalidixic acid; STR, streptomycin; SXT, trimethoprim–sulfamethoxazole; TET, tetracycline; TIO, ceftiofur.
Based on the results of the antibiotic sensitivity assay, both resistant (n = 7) and susceptible (n = 21) isolates were randomly selected for genome sequencing and analysis (Table 1). The genomes of 28 E. coli isolates were sequenced on a NextSeq 500 (Illumina, San Diego, CA). DNA was extracted using the QIAamp DNA Mini Kit on a QIACube DNA Extraction platform (QIAGEN, Valencia, CA), and sequencing libraries were created using the Nextera XT Library Kit (Illumina). Raw sequencing reads were curated for quality and length using Trimmomatic v. 0.36 (LEADING:20 TRAILING:20 SLIDINGWINDOW:4:20 MINLEN:36) (Bolger et al., 2014) and Deconseq v.0.4.3 (Schmieder and Edwards, 2011) and were assembled into draft genomes using SPAdes 3.13.0 (Bankevich et al., 2012). Contigs <200 bp were trimmed from each genome. The draft genomes were analyzed for sequence types (STs), ST complexes (ST Cplxs), and O and H antigens, and the presence/absence of transferrable antibiotic resistance genes (ARGs) was determined using MLST 2.0, SerotypeFinder 2.0, and ResFinder 4.0 hosted by the Center for Genomic Epidemiology webserver (Larsen et al., 2012; Joensen et al., 2015; Bortolaia et al., 2020; Thomsen et al., 2016). Novel STs were identified by EnteroBase (Alikhan et al., 2018; Achtman et al., 2020). Phylogenetic groups were determined in silico (Beghain et al., 2018). The presence of virulence genes was determined using a combination of VirulenceFinder 2.0 and BLASTN (percent identity ≥90%, query coverage = 100%, e-value = 10E-30) (Joensen et al., 2014; Thomsen et al., 2016). Core genome single nucleotide polymorphisms (SNPs) of the study genomes (ARS-CC) along with representatives of the phylogenetic groups (Supplementary Table S1) were identified using the Harvest package (Treangen et al., 2014). These SNPs were then used to infer a maximum likelihood phylogenetic tree using RAxML with default settings and 1000 bootstrap replicates (Stamatakis, 2014). All genome sequencing data generated in this study have been deposited and are publicly available in National Center for Biotechnology Information (NCBI) under BioProject ID PRJNA563910. Institutional animal care and use committee review was not required since fecal samples were collected from the pen floors with no manipulation of the animals.
Results
The results of the genomic analysis are summarized in Table 2. Only three phylogenetic groups were identified among the 28 isolates, with B1 being the most frequently detected (n = 23), followed by A (n = 4), and a single isolate was identified as either a member of phylogenetic group E or Cryptic (identified in silico as both). The maximum likelihood phylogenetic analysis demonstrated a greater degree of phylogenetic diversity than did the in silico phylogenetic grouping analysis (Fig. 1). However, multiple strains from the same locations were grouped together indicating that they were clonal isolates of the same strains that may have been predominant in these animals/locations. Isolate ARS-CC10126 clustered with phylogenetic group E references genomes in the phylogenetic analysis indicating that it is not a member of the Clade I group (Fig. 1).
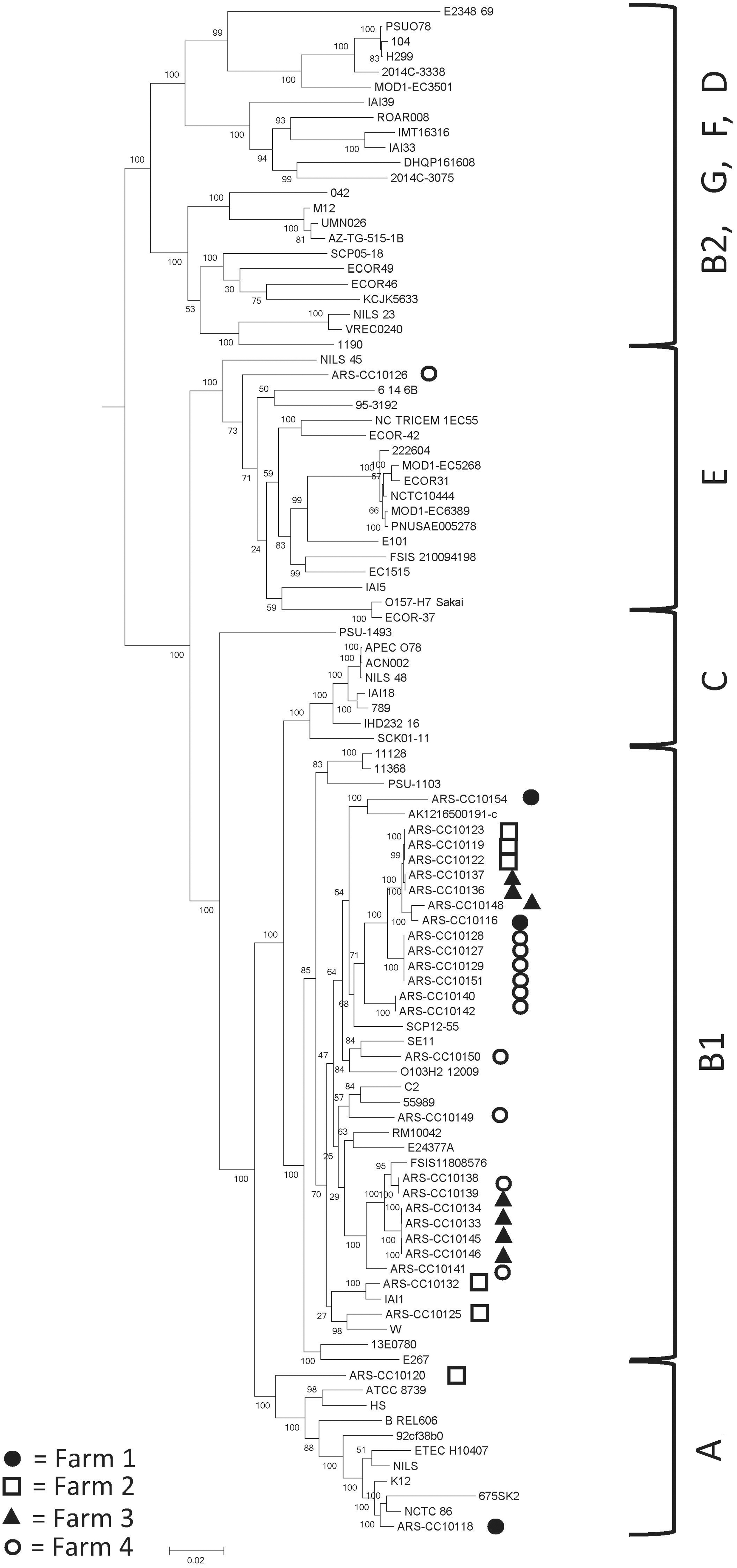
Maximum likelihood phylogenetic tree of Escherichia coli representing the major phylogenetic groups (Supplementary Table 1), including 28 isolated from bovine sources in Mozambique (with the prefix “ARS-CC”), with 1000 bootstrap replicates. Scale bar represents number of substitutions per site. Black circles, Farm 1; white squares (black outline), Farm 2; black triangles, Farm 3; white circles (black outline), Farm 4. Phylogenetic groups are noted on the right side of the figure.
Genomic Data for Each Isolate
Isolate ID, phylogenetic groups, ST, ST Cplx, transferrable antibiotic resistance genes, antibiotic resistance conferring mutations (SNPs), plasmid replicons, and O- and H-antigens.
ST, sequence type; ST Cplx, sequence type complex.
A total of 16 STs were identified among the isolates (Table 2), with ST58 being the most frequently identified (5 genomes) followed by ST642 and ST223 (4 genomes each). Three previously unidentified STs (ST9214, ST9215, and ST9216) were identified. Five ST Cplxs were identified, with ST155 Cplx being the most frequently detected (11 genomes), followed by ST278 Cplx (6 genomes), and one isolate of ST10 Cplx, ST23 Cplx, and ST469 Cplx. Eight isolates were assigned to seven singleton complexes (ARS-CC10120, ARS-CC10125, ARS-CC10126, ARS-CC10140, ARS-CC10141, ARS-CC10142, ARS-CC10149, ARS-CC10154) (Table 2).
The serotypes of the isolates were determined in silico using SerotypeFinder. The O-antigen coding sequences were detected and typed in 15 genomes, whereas the H-antigen coding sequences were detected and typed in all genomes. The complete serotypes detected were O1:H21, O162:H48, O162:H7, O182:H34, O187:H52, O18ac:H21, O6:H21, O8:H19, and O89/O162:H4. The most frequently detected H-antigen was H21 (10 genomes), followed by H52 and H7 (4 genomes each).
Of the seven resistant isolates, four were resistant to at least four antibiotics, whereas three were only resistant to tetracycline (Table 1). All resistant isolates encoded ARGs that correspond with the results of the phenotypic antibiotic sensitivity assays (Table 2). Seven different ARGs were detected, and the tetracycline resistance gene tet(B) was the most common (7), followed by aminoglycoside resistance genes aph(3″)-Ib and aph(6)-Id and sulfonamide resistance gene sul2 (4 each). The aminoglycoside, β-lactam, and trimethoprim resistance genes, aadA1, bla TEM-1B, and dfrA1, respectively, were each detected once in the same genome. β-Lactam-conferring point mutations in the ampC promoter (−42C>T) (Caroff et al., 1999) were identified in three isolates that were resistant to amoxicillin/clavulanic acid and ampicillin, and these isolates had increased minimum inhibitory concentrations to cefoxitin (one of them being resistant to cefoxitin).
In ARS-CC10118, the aph(3″)-Ib, aph(6)-Id, and sul2 genes were flanked by IS6-like element IS26-family transposase sequences and located on a 4539 bp contig that shows >95% similarity to several E. coli plasmid and chromosomal sequences. The dfrA1, aadA1, aph(3″)-Ib, and aph(6)-Id resistance genes were located on a 21 kb contigs that is highly similar to E. coli chromosomal sequences. In isolates ARS-CC10119, ARS-CC10122, ARS-CC10123, the aph(3″)-Ib, aph(6)-Id, and sul2 genes were flanked by an IS91-family transposase and an IS4-like element ISVsa5-family transposase, and these contigs were highly similar to chromosomal E. coli sequences.
Multidrug-resistant (MDR; resistant to ≥3 classes of antibiotics) isolates were either ST10 or ST58. The MDR ST10 isolate ARS-CC10118 harbored IncFII, IncFIA, IncFIB, and IncQ plasmid replicons. All ST58 isolates harbored plasmids with IncFIC and IncFIB replicons, but there were no replicons that were uniquely detected in the MDR isolates or pan-susceptible isolates. IncFIB, IncFII, and IncFIC were the most frequently detected plasmid replicons (n = 25, 18, and 18, respectively), followed by Col156 and IncI1, both of which were detected in nine genomes.
In total, 51 different virulence genes were detected at least once among the genomes (Fig. 2), and 25 virulence genes were detected in every genome. Thirteen isolates encoded the heat-stable toxin astA. Twenty-five, 25, and 16 genomes encoded the lpfA, gad, and iss genes, respectively. The highest number of virulence genes detected in one isolate was 47 (ARS-CC10126). This isolate encoded the chu heme utilization operon, the air adhesin, and the eilA regulator. The fewest virulence genes detected in a single isolate was 28. The median number of virulence genes was 37. Shiga-toxin genes (stx), intimin (eae), translocated intimin receptor (tir), and bundle-forming pilus (bfp) were not identified in any of the genomes.

Presence/absence heat map of virulence factors that were detected in at least one genome. Black, present; white, absent.
Discussion
Antibiotic resistance remains a significant global threat, but the presence of these organisms in food-producing animals remains understudied in many parts of the world. To better assess and address this issue, more data need to be acquired on the presence and types of antibiotic-resistant bacteria in many understudied regions. Recent work in Mozambique has focused on resistant bacteria associated with human clinical infections (Mandomando et al., 2009, Mandomando et al., 2010, Ibarz-Pavón et al., 2011, Mashana et al., 2013, Berendes et al., 2019). However, the presence and types of these bacteria in food production animals have not been adequately described.
This work represents the first study on the genomic characteristics of both antibiotic-resistant and -susceptible E. coli in bovine feces in and around Maputo, Mozambique. The results of this study further demonstrate the global presence of genes conferring resistance to aminoglycosides, tetracycline, sulfonamides, and β-lactams, which have been frequently identified in E. coli in other parts of the world. Of interest is the presence of the β-lactamase gene, bla TEM-1B, and strains with enhanced β-lactamase-conferring mutations in the ampC-promoter. β-lactams are among the most prescribed antibiotics worldwide (WHO, 2018), and the presence of bla TEM-1B and AmpC chromosomal β-lactamases in bovine feces indicates that resistance to these antibiotics is present within the bacterial populations of food animals in Mozambique. However, other β-lactamases, such as CTX-M, OXA, and SHV, were not identified among the isolates. CTX-M and SHV genes have been detected in Gram-negative bacteria isolated from human extraintestinal infections in Mozambique, demonstrating that they are present in the region, and are circulating within the human population (Pons et al., 2015; Guiral et al., 2018). The presence of the trimethoprim resistance gene, dfrA1, is significant from a human health perspective. This antibiotic is a broad-spectrum antimicrobial agent active against enteric pathogens such as E. coli/Shigella and Salmonella enterica, and trimethoprim–sulfamethoxazole (SXT) resistance has become common in human clinical S. enterica and E. coli isolated from pregnant women in Mozambique (Mandomando et al., 2009; Sáez-López et al., 2016). SXT is extensively used as a treatment for opportunistic infections associated with HIV in parts of Mozambique, indicating that the presence of this SXT resistance represents a potential public health threat in this country (Sáez-López et al., 2016).
The E. coli in this study were not highly diverse with respect to phylogenetic groups/lineages, but they are approximately similar to E. coli isolated from bovine sources worldwide, which were shown to belong predominantly to phylogenetic group B1 (Carlos et al., 2010). Within phylogenetic group B1, there were multiple STs detected. Interestingly, several of the STs identified in this study, such as ST10 and ST58, are considered globally disseminated, whereas three novel STs were identified. This suggests that there may be a significant number of unrecognized/new E. coli sublineages circulating among the bovine populations in this region that have not yet been described.
Several E. coli belong to STs that are associated with human infections. Among these was an ST10 (O89/O162:H4-A-ST10) isolate, which was MDR and encoded genes known to confer resistance to aminoglycosides, β-lactams, sulfonamides, tetracycline, and trimethoprim. ST10 isolates have been isolated from human gastrointestinal and urinary tract infections and are considered globally distributed, which is further confirmed by the results of our study (Manges et al., 2015). Five ST58 (ST155 Cplx) isolates and six other isolates of three STs belonging to the ST155 Cplx were identified as well. Of these, ST58 and ST223 have been repeatedly isolated from human clinical infections (McKinnon et al., 2018). ST223 strains belonged to the O18ac:H21 serogroup that was previously reported as invasive and enteropathogenic to piglets (Wada et al., 2004). ST683 appears to be restricted mainly to livestock and poultry, and ST9216 is a novel ST described first in this study (Schlager et al., 2008; Zhou et al., 2019). ST9216 and ST155 strains are the most closely related and share six of seven alleles. ST162 of the ST469 Cplx was isolated once and has also been recovered from human clinical infections (Zhou et al., 2019). The remaining STs have not been isolated from human clinical cases or have been infrequently isolated from such cases. Interestingly, the ST encoding the most virulence factors, ST9215, has not been previously described. Based on the MLST alleles, this strain is most closely related to an ST8826 isolate (PapRG-04-4) that was isolated in 2016 from a wild animal in the Gambia, over 6000 km away from Mozambique (one allele difference). This further suggests that there may be unrecognized/new lineages of closely related strains circulating in this continent that may be revealed by broad and extensive sampling of nonhuman matrices in understudied regions. Multiple virulence factors involved in severe intestinal and gastrointestinal infections were identified among the isolates, including astA and the heme utilization operon. We did not detect genes encoding Shiga toxins, although Shiga-toxigenic isolates are infrequently identified compared with non-toxigenic isolates. Results of this study indicate that there is a potential for these bovine E. coli isolates to cause human infections.
There are multiple reservoirs of antibiotic-resistant bacteria that are known to harbor pathogens that cause mild-to-severe infections in humans. Identifying resistant bacteria in clinical human infections as part of an outbreak investigation helps to determine the exact causative agents of intractable human infections. However, it is also essential to identify those strains (resistant and susceptible) that are circulating in the animal populations that serve as food sources and/or companion animals for humans, as well as those animals living near human populations. In-depth analyses of these strains and sources may help identify potential reservoirs of the causative agents of human disease and allow for potential mitigation of these public health risks. These sources and strains are not well defined in many parts of the world, and this represents a concerning limitation of our understanding of resistant bacteria that may be transmitted to humans.
Results of this study indicate that antibiotic-resistant E. coli are present in bovine sources in Mozambique. This work leveraged a composite sampling strategy, which provides information on herd-level carriage of resistant and susceptible bacteria (Lombard et al., 2012), but future work should also address within-herd abundance of these bacteria and the management pressures that may be associated with carriage of resistance. This will help to more adequately describe the true diversity of drug-resistant bacteria in these animals and help assess the health risk of resistant E. coli to those individuals interacting with these animals or using them as sources of food. Future work should assess the virulence potential of more antibiotic-resistant and -susceptible E. coli strains from a broader geographic region over time and compare these with strains isolated from humans to more adequately assess the circulation of these strains between animals and humans in Mozambique.
Footnotes
Acknowledgments
We gratefully acknowledge the assistance of Laura Del Collo and Jakeitha Sonnier (Agricultural Research Service, U.S. Department of Agriculture [USDA]), and Dr. Jose Fafetine (Center of Biotechnology, University Eduardo Mondlane). The mention of a trade name, proprietary product, or specific equipment does not constitute a guarantee or warranty by the USDA and does not imply approval to the exclusion of other products that might be suitable.
Disclosure Statement
No competing financial interests exist.
Funding Information
A.M. and E.T. were supported by Italian Agency for Cooperation Development funds, Project AID11096.
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.