
Abstract
Salmonella spp. are among the most prevalent foodborne pathogens. Rapid identification of etiologic agents during foodborne outbreaks is of great importance. In this study, we report a traceback investigation of a Salmonella outbreak in China. Metagenomic sequencing of suspected food samples was performed on MinION and MiSeq platforms. Real-time nanopore sequencing analysis identified reads belonging to the Enterobacteriaceae family. MiSeq sequencing identified 63 reads specifically mapped to Salmonella. Conventional methods including quantitative-PCR and culture-based isolation confirmed as Salmonella enterica serovar Typhimurium. The foodborne outbreak of Salmonella Typhimurium was further recognized by whole-genome sequencing and pulsed-field gel electrophoresis analysis. Our study demonstrates the ability of metagenomic sequencing to rapidly identify enteric pathogens directly from food samples. These results highlight the capacity of metagenomic sequencing to deliver actionable information rapidly and to expedite the tracing and identification of etiologic agents during foodborne outbreaks.
Introduction
Salmonella spp. are important foodborne pathogens that cause ∼93.8 million cases of gastroenteritis annually, thus posing serious threats to global health (Majowicz et al., 2010). In China, Salmonella spp. are responsible for 70–80% of bacterial gastroenteritis cases (Zhan et al., 2017), cause substantial socioeconomic burdens (Liu et al., 2018), and are widely distributed, which can be detected from various sources, including food catering workers (Xu et al., 2019), retail foods (Chen et al., 2019), and chickens in large breeder farms (Yang et al., 2019). Salmonella enterica serovar Typhimurium is one of the most common serotypes detected in retail foods in China (Wang et al., 2017). In addition, the increasing prevalence of multidrug resistance among Salmonella spp. poses a formidable challenge to treatment (Xiang et al., 2020).
Timely identification of enteric pathogens is critical for the prevention and control of foodborne outbreaks. The conventional culture-based isolation method is time-consuming and usually requires several days for pathogen identification, subtyping, and antibiotic susceptibility testing. A variety of methods have been applied for the rapid detection of foodborne pathogens (Law et al., 2014; Zhao et al., 2019). Whole-genome sequencing can increase subtyping resolution, provide epidemiology evidence, and has been applied for outbreak investigation and surveillance in China (Chen et al., 2020; Xiang et al., 2020; Jiang et al., 2020; Li et al., 2021) and other countries (Nouws et al., 2020; Smith et al., 2020; Vaughn et al., 2020).
Metagenomic next-generation sequencing (mNGS) allows for culture-independent analysis, higher resolution subtyping of foodborne bacteria, and rapid identification of contaminated food during outbreaks. Therefore, mNGS constitutes a powerful tool for the early detection of pathogens and investigation of foodborne outbreaks (Kim et al., 2018; Grützke et al., 2019; Buytaers et al., 2021). However, because of its limited accessibility, high economic costs, and relatively long turnaround time, mNGS has been used primarily as an adjunct to culture based-detection to identify food sources of outbreaks.
On-site direct sequencing of samples during foodborne outbreaks in real time could shorten the duration of empiric therapy, expedite rapid diagnosis and optimal clinical management. With the portable MinION sequencer, Nanopore sequencing can detect possible pathogens in real time with a turnaround time of <6 h (Greninger et al., 2015; Charalampous et al., 2019), but has lower accuracy and sequencing throughput compared with other NGS platforms (Rang et al., 2018; Wilson et al., 2019). Nanopore sequencing has been successfully applied for Salmonella serotype prediction from bacterial isolates (Quick et al., 2015; Xu et al., 2020). However, the application of direct metagenomic sequencing to uncultured food samples remains challenging and requires further investigation.
In this study, we reported a foodborne outbreak of diarrhea from a canteen in Beijing in August 2018. Culture-independent metagenomic sequencing using MinION and MiSeq platforms was applied to samples of salted duck eggs, which were implicated by the epidemiological investigation as the possible source. Salmonella Typhimurium was confirmed as the pathogen by culture-based isolation and real-time quantitative-PCR. Our study highlights the utility of metagenomic sequencing for pathogen identification directly from food samples during foodborne outbreaks.
Materials and Methods
Sample collection and genomic DNA extraction
Anal swab samples were collected from 13 patients with diarrhea and stirred in 500 μL phosphate-buffered saline (PBS). Sterile cotton swabs were brushed over the surface of all the remaining salted duck eggs and 51 other reserved food samples in the canteen and fully stirred in 500 μL PBS. Genomic DNA was extracted directly from the buffer using the TIANGEN DNA/RNA isolation kit (TIANGEN, Beijing, China) according to the manufacturer's instructions. All of the samples used in this study were collected through routine surveillance as no personally identifiable data were included. The ethics of the study was reviewed and supervised by Chinese PLA Center for Disease Control and Prevention.
Real-time quantitative PCR identification, pathogen isolation, and pulsed-field gel electrophoresis analysis
Real-time quantitative PCR assays were used to screen for suspected pathogens, including norovirus, rotavirus, astrovirus, enteric adenoviruses, Vibrio cholerae, Vibrio parahaemolyticus, Salmonella spp., Shigella spp., and enteropathogenic Escherichia coli. The specimens were also cultured on salmonella-shigella agar (Land Bridge Technology, Beijing, China) at 37°C for 24 h. Bacterial colonies were isolated and identified by the GEN III Omnilog microbial identification system using GEN III Microplate™ test panel (Biolog, CA, USA) according to the manufacturer's instructions.
Cell density was adjusted in the range of 90–98% turbidity with the turbidimeter and the 100 μL cell suspension was added into all wells. The Microplate was then incubated for up to 24 h and results were interpreted by the system. All five Salmonella Typhimurium isolates were analyzed by pulsed-field gel electrophoresis (PFGE) as described previously (Xie et al., 2015). DNA was digested by XbaI restriction enzyme (Takara Bio, Shiga, Japan) and separated using a CHEF Mapper PFGE system (Bio-Rad, CA, USA) with Salmonella serotype Braenderup strain H9812 as a reference size standard.
Nanopore sequencing of the food sample
Genomic DNA extracted from salted duck eggs was quantified using the Qubit 3.0 fluorometer (Thermo Fisher Scientific, CA, USA) and used directly for MinION sequencing. A library was prepared using a rapid sequencing kit (Oxford Nanopore Technologies, Cambridge, United Kingdom) and loaded onto a FLO-MIN106 R9.4 Flow Cell according to the manufacturer's protocol. Sequencing of the sample was performed using MinKNOW software.
Illumina MiSeq sequencing of the food sample and bacteria cultures
Genomic DNA of five cultured Salmonella Typhimurium isolates was extracted for whole-genome sequencing using the High Pure PCR Template Preparation Kit (Roche, Basel, Switzerland) according to the manufacturer's protocol. A library of the salted duck egg food sample and the five cultured Salmonella Typhimurium isolates was prepared using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England BioLabs, CA, USA) following the manufacturer's guidelines. Sequencing was then performed on MiSeq (Illumina, CA, USA) with a pair-end 300 bp strategy.
Metagenomic and phylogenetic analysis
Metagenomic sequence classification of the salted duck egg sample was performed using Kraken (v1.1.1) with default parameters and a standard database constructed from complete bacterial, archaeal, and viral genomes in Refseq (Wood and Salzberg, 2014). The raw reads were mapped to the reference genome of Salmonella Typhimurium LT2 using BWA (v0.7.12) (Li and Durbin, 2009).
Draft genomes of the five isolates SBJ0C3, SBJ0E7, SBJ06P, SBJ09P, and SBJ011P were assembled de novo using SPAdes (v3.6.2) with Kmers set as 21, 33, and 55 (Bankevich et al., 2012). Complete genomes of 38 Salmonella Typhimurium strains from China were downloaded from the National Center for Biotechnology Information database for phylogenetic analysis. Single nucleotide polymorphisms were identified using kSNP3.0 with calculated loci set as “-core” (Gardner et al., 2015). A Maximum-Likelihood phylogenetic tree was constructed using RAxML (v8.2.4) with the general time-reversible model and a gamma distribution based on 1000 bootstraps (Stamatakis, 2014).
The phylogenetic tree was visualized using iTOL (v6) (Letunic and Bork, 2021). Antimicrobial resistance genes were identified by aligning genome sequences against the Comprehensive Antibiotic Resistance Database (McArthur et al., 2013) using BLASTn (v.2.2.28) (Camacho et al., 2009). Average nucleotide identity was calculated using the JSpeciesWS webserver (Richter et al., 2016). Sequence types were determined through the multilocus sequence typing (MLST) webserver (Larsen et al., 2012).
Data availability
The GenBank accession numbers for the sequences of outbreak isolates SBJ0C3, SBJ0E7, SBJ06P, SBJ09P, and SBJ011P are JAHIBJ000000000, JAHIBI000000000, JAHIBG000000000, JAHIBF000000000, and JAHIBH000000000, respectively.
Results
Epidemiology investigation, quantitative-PCR screening, and isolate identification
A foodborne outbreak was reported in August 2018, in Beijing. Thirteen patients all showed symptoms of diarrhea and some patients also showed fever, abdominal pain, or vomiting after taking a meal in a canteen. Epidemiology investigation revealed that most of the patients had eaten the salted duck eggs supplied by the same grocery shop at breakfast in the same day. The remaining salted eggs and 51 other food samples were collected for further analysis. In addition, anal swab samples were obtained from all the patients.
All the samples were used for pathogen isolation and quantitative-PCR screening. Quantitative-PCR screening identified Salmonella in 5 patient specimens and 16 food samples. Culture-based isolation yielded five isolates, two from food samples (Isolate SBJ0C3: salted duck egg, Isolate SBJ0E7: scrambled eggs) and three others from anal swabs (Isolates SBJ06P, SBJ09P and SBJ011P). All five isolates were identified as Salmonella Typhimurium according to the biochemical test by the GEN III Omnilog microbial identification system (Supplementary Table S1). The salted eggs purchased from the same vendor were collected for quantitative-PCR screening and pathogen isolation, but neither of the results was positive.
Metagenomic sequencing of the salted duck egg sample with MinION and MiSeq platform
Consequent to the results of the epidemiological investigation, metagenomic sequencing for rapid pathogen identification was performed first on the salted egg sample. The sample had a DNA concentration of 1.87 ng/μL and was sequenced both on Nanopore MinION and Illumina MiSeq platforms. MinION generated 15.64 k reads with a ∼6 h run. By assessing a quality cutoff (Qscore >7), a total of 5743 reads were selected for subsequent analysis with an average length of 1554 bp (Supplementary Fig. S1). MiSeq generated ∼1.84 M reads with a Q30 value of 89.93%.
Taxonomic analysis using Kraken revealed that bacterial reads accounted for 16.6% in all nanopore reads. The MinION platform yielded 108 reads that were specifically matched with Pseudomonas fulva and 41 reads specifically mapped to Stenotrophomonas maltophilia. However, no specific reads mapped to the Salmonella genus were identified. Reads mapped with Enterobacteriaceae were detected by nanopore sequencing within 1 h. The number of Enterobacteriaceae-matched reads did not increase after 4 h of sequencing (Supplementary Fig. S2). Taxonomic analysis indicated that 18.2% of MiSeq reads were identified as having bacterial origin. MiSeq generated 63 reads, which were specifically aligned to Salmonella. All reads were subsequently mapped to the reference genome Salmonella Typhimurium LT2 (AE006468.2) and showed 0.75 × average depth and 19.7% coverage (Supplementary Fig. S3).
The MiSeq metagenomic data contained 243 families, whereas the MinION data disclosed 49 families. Seventeen Enterobacteriaceae genera were identified by the MiSeq platform, compared with eight in the MinION data. However, the read proportions of microbial populations identified by the two platforms were generally similar (Fig. 1). 5.0% of the bacterial reads were identified as Enterobacteriaceae in MiSeq data in comparison with 5.6% for MinION. Such consistence was likewise observed for Pseudomonadaceae (44.9% of MinION bacterial reads and 52.2% of MiSeq bacterial reads) and Xanthomonadaceae (7.9% and 9.1%, respectively).
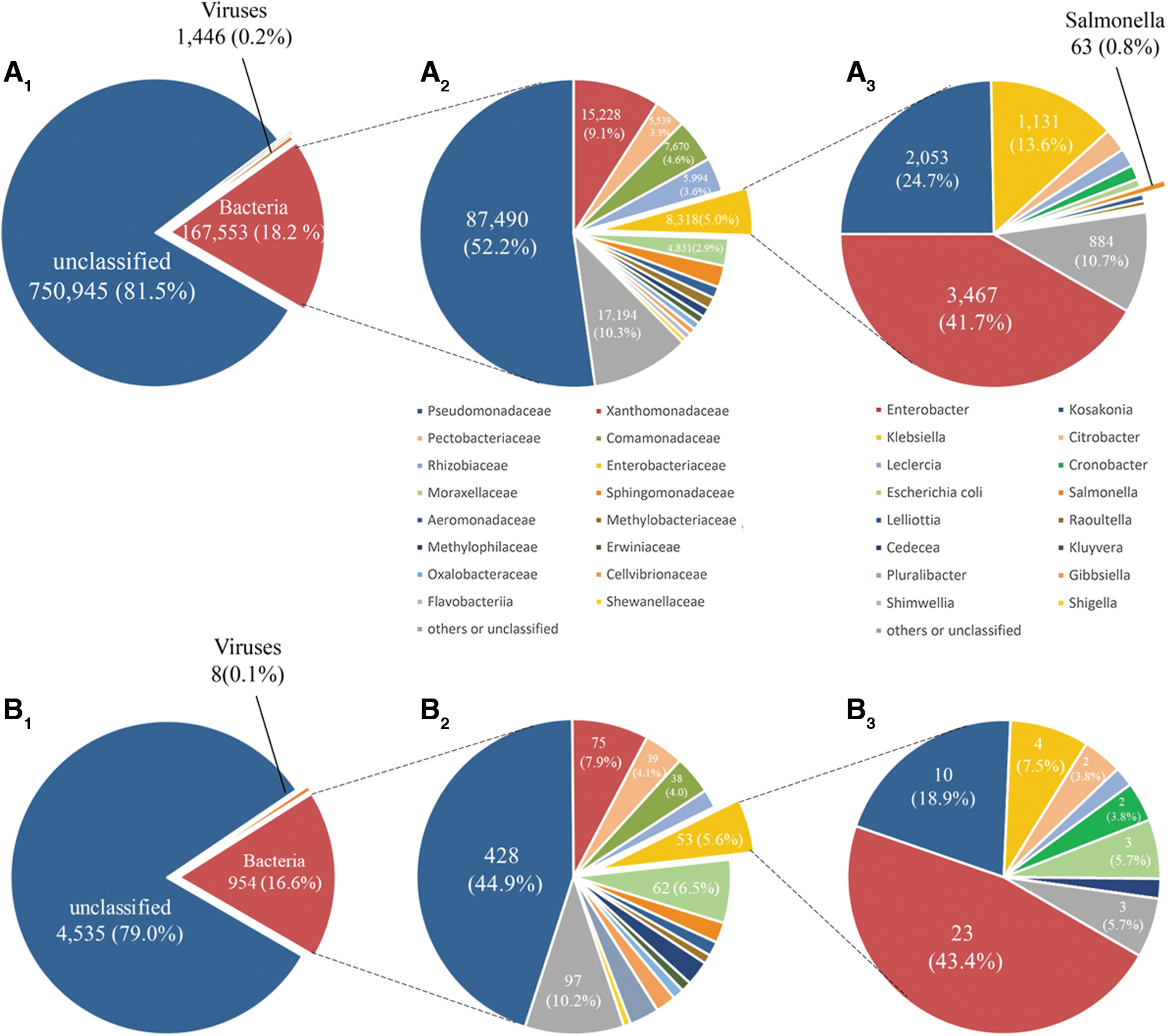
Distribution of microbial diversity from MiSeq and MinION sequencing.
Genetic relatedness of the five Salmonella Typhimurium isolates
To investigate the genetic relatedness of the five isolates, molecular typing and whole-genome sequencing were performed to investigate the relationship of the five Salmonella Typhimurium isolates. MLST analysis showed that all five isolates belonged to sequence type 34. The isolates also shared the same PFGE pattern (Supplementary Fig. S4).
Genome sequences were assembled based on whole-genome sequencing (Supplementary Table S2) and the average nucleotide identities ranged from 99.82% to 99.98%. Phylogenetic analysis with other Salmonella Typhimurium isolates from China (Supplementary Table S3) revealed that the five isolates were clustered together and had a close relationship with strain WW012 from a 2014 ready-to-eat pork sample from China (Fig. 2). These results indicated that the five outbreak isolates may have originated from a single clone.
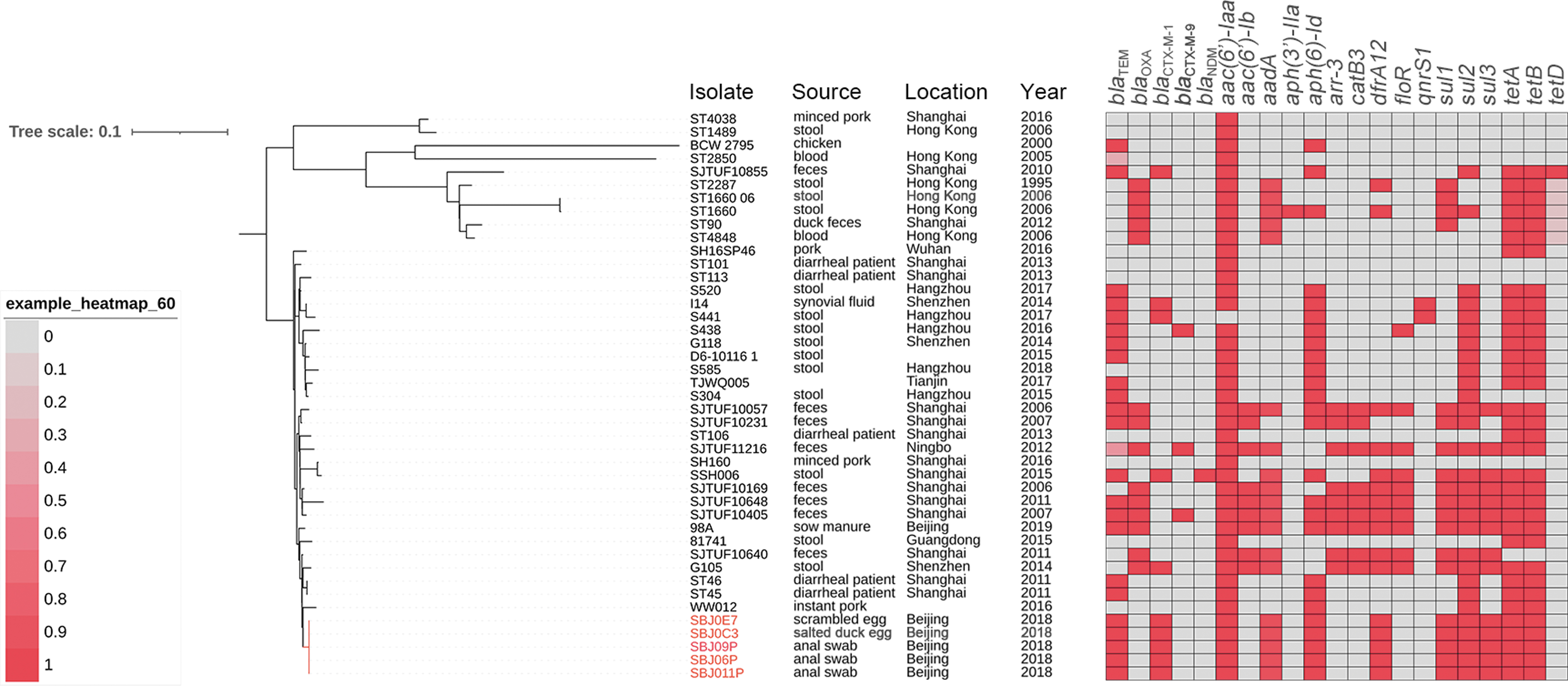
Phylogenetic tree and antimicrobial resistance gene profiles of the outbreak and other Salmonella Typhimurium isolates from China.
Discussion
We report the application of MiSeq and MinION metagenomic sequencing for pathogen identification in a Salmonella foodborne outbreak in China and our results suggest that direct metagenomic sequencing has great potential to rapidly identify etiologic agents from uncultured samples during foodborne outbreaks.
Four strategies were performed for pathogen detection in this study. Real-time quantitative PCR assay has the shortest turnaround time (∼5 h), whereas pathogen isolation takes days (∼60 h) to obtain the results. Both of them required priori knowledge of pathogens to design specific primers or culture conditions. mNGS can obtain all microbes from one sample. In this study, MiSeq sequencing identified more species than MinION sequencing including Salmonella Typhimurium and showed higher sensitivity of pathogen detection.
In contrast, MinION generated data in real time and has a shorter turnaround time than MiSeq (∼8.5 and ∼70 h, respectively). Comparison of the four methods (Supplementary Table S4) revealed that nanopore sequencing had short turnaround time but was limited by its low throughput. The combination of different sequencing platforms with traditional methods could provide more information and help the identification of the possible pathogen.
Our study had several limitations. As the eggs implicated in the epidemiological investigation were cooked and vacuum-packed, contamination may have occurred during factory packaging or canteen processing. However, not all of the food selections sold in the canteen were reserved. After the Salmonella cases were reported, the canteen was cleaned; consequently, samples from chefs, knives, and kitchen environments could not be obtained. The limited data from the outbreak investigation confounded the identification of the sources of salted egg contamination. In addition, salted eggs purchased from the same vendor did not yield Salmonella, which impeded the tracing of the source of contamination. The salted duck eggs were likely to be cross-contaminated during food processing.
The portable MinION sequencer showed great potential for enabling on-site pathogen detection during infectious disease outbreaks. However, the sensitivity and specificity were impeded by sequencing throughput and the pathogen abundance in the sample. In this study, both MiSeq and MinION platforms identified the major bacterial constituents of uncultured samples, but MinION did not identify specific Salmonella reads possibly due to the low data yield.
The recommended gDNA content for the rapid sequencing kit is 400 ng, and the low input of samples may result in low output of sequencing data. Low bacterial loads of enteric pathogens in food samples pose major obstacles to the application of nanopore sequencing in foodborne outbreak investigations. Effective enrichment methods are urgently needed to improve the sensitivity of metagenomic sequencing of culture-independent samples.
Most published studies have used PCR-enriched samples for nanopore sequencing (Quick et al., 2017; Stubbs et al., 2020). Furthermore, a variety of metagenomic sequencing variables, including sample type, pathogen content, and extraction method should be considered and optimized to improve the efficiency of microbiologic diagnosis. Even though mNGS is a powerful approach to detect the unknown pathogens, it is often difficult to directly identify the causative agent alone.
Many point-of-care methods for foodborne pathogen detection, such as microfluidic devices (Han et al., 2021), zinc oxide-based sub-micro pillar array (Lee et al., 2020), bacterial capturing nanotopographical trap (Kim et al., 2021), smartphone-integrated paper sensing system (Wang et al., 2020a), and lateral flow strip (Wang et al., 2020b), have been developed. These methods can provide rapid, sensitive, and on-site detection, but need priori knowledge of the pathogens. Combining mNGS and other point-of-care assays would achieve more accurate detection and quantitation of pathogens.
Conclusions
Foodborne outbreaks pose major threats to public health. Our results demonstrate that metagenomic sequencing directly from uncultured food samples is feasible for the identification of foodborne pathogens. Nanopore sequencing was restricted by throughput and was not applicable for samples with low microbial abundance. In contrast, MiSeq sequencing was able to reveal comprehensive microbial populations, showing higher sensitivity of pathogen detection. Molecular typing and whole-genome sequencing revealed Salmonella Typhimurium clonal expansion. The identification of salted duck eggs as the possible intermediate reservoir of the Salmonella suggests that greater care is needed during the preparation and handling of this particular ready-to-eat food.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
The study was supported by grants from the Mega-projects of Science and Technology Research (No. 2017ZX10303405-003), the Beijing Natural Science Foundation (No. 5172029), and the Beijing Noval program (No. Z181100006218110).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.