
Abstract
Enterohemorrhagic Escherichia coli (EHEC) is one of the most common E. coli pathotypes reported to cause several outbreaks of foodborne illnesses. EHEC is a zoonotic pathogen, and ruminants, especially cattle, are considered important reservoirs for the most common EHEC serotype, E. coli O157:H7. Humans are infected indirectly through the consumption of food (milk, meat, leafy vegetables, and fruits) and water contaminated by animal feces or direct contact with carrier animals or humans. E. coli O157:H7 is one of the most frequently reported causes of foodborne illnesses in developed countries. It employs two essential virulence mechanisms to trigger damage to the host. These are the development of attaching and effacing (AE) phenotypes on the intestinal mucosa of the host and the production of Shiga toxin (Stx) that causes hemorrhagic colitis and hemolytic uremic syndrome. The AE phenotype is controlled by the pathogenicity island, the locus of enterocyte effacement (LEE). The induction of both AE and Stx is under strict and highly complex regulatory mechanisms. Thus, a good understanding of these mechanisms, major proteins expressed, and environmental cues involved in the regulation of the expression of the virulence genes is vital to finding a method to control the colonization of reservoir hosts, especially cattle, and disease development in humans. This review is a concise account of the current state of knowledge of virulence gene regulation in the LEE-positive EHEC.
Introduction
Escherichia coli is a Gram-negative, rod-shaped, and facultative anaerobic bacteria in the family of Enterobacteriaceae (Bahrani-Mougeot, 2009). The bacterium was first identified by Theodor Escherich in 1885 (Shulman et al., 2007). Most E. coli strains are commensals inhabiting the lower intestinal tract of humans and warm-blooded animals (Backhed et al., 2015; Dicks et al., 2018; Ramos et al., 2020). The commensal E. coli is part of normal intestinal microbiota that coexist with their host in a mutually symbiotic relationship (Walter and Ley, 2011).
Normal microbiota benefits the host in digesting nutrients, producing vitamins, preventing pathogens colonization, and inducing the development and maintenance of normal adaptive immune responses (Mellies and Barron, 2006; Kau et al., 2011; Kamada et al., 2013; Belkaid and Hand, 2014; Mellies and Lorenzen, 2014). However, the host's intestinal tract serves as a natural habitat and food source for normal microbiota growth and survival.
Pathogenic E. coli are categorized into seven intestinal and two extraintestinal pathotypes (Nash et al., 2010; Liu et al., 2020). The intestinal pathotypes are enterohemorrhagic E. coli (EHEC), enteropathogenic E. coli (EPEC), enteroinvasive E. coli, enteroaggregative E. coli, enterotoxigenic E. coli, diffusely adherent E. coli (Nataro and Kaper, 1998; Kaper et al., 2004), and adherent-invasive E. coli associated with Crohn's disease (Martinez-Medina et al., 2011; Rahmouni et al., 2018; Camprubí-Font et al., 2019; Lee et al., 2019; Mayorgas et al., 2021). The extraintestinal pathotypes or extraintestinal pathogenic E. coli includes uropathogenic E. coli (Chen et al., 2006; Schwartz et al., 2011) and newborn meningitis-associated E. coli (Logue et al., 2012; Wijetunge et al., 2015).
Shiga toxin (Stx) was originally identified in 1977 (Konowalchuk et al., 1977) for its verocytotoxicity and later considered a Shiga-like toxin due to its similarity with the Shiga toxin of Shigella dysenteriae type 1 (O'Brien et al., 1982; Nataro and Kaper, 1998). In addition to E. coli O157:H7, most EHEC infections are caused by four O serogroups O26, O103, O111, and O145 (Eichhorn et al., 2015). E. coli O157:H7, the most common strain of EHEC, was first reported in the United States in 1982 (CDC, 1982). Most EHEC-associated outbreaks are foodborne (CDC, 2009).
EHEC colonizes the intestinal tract of animals, mainly ruminants, and cattle are the main reservoirs for EHEC O157:H7 (Pruimboom-Brees et al., 2000; Besser et al., 2001; Naylor et al., 2003; Gyles, 2007; Nguyen and Sperandio, 2012).
Most EHEC infections are acquired by direct contact with ruminant feces or indirectly through contaminated foods such as beef, milk, apple juice (Caprioli et al., 2005; Greig and Ravel, 2009; Ferens and Hovde, 2011; Cramer, 2014; Posada-Izquierdo et al., 2016; van Hoek et al., 2019; Pakbin et al., 2021), fruits, leafy vegetables (Doyle and Erickson, 2008; Sodha et al., 2011; Beutin and Martin, 2012; Thao et al., 2019), and water (Olsen et al., 2002).
EHEC produces different virulence factors and Shiga toxin (Kaper et al., 2004), which are responsible for the development of attaching and effacing (AE) phenotypes on the host intestinal mucosa, hemorrhagic colitis (HC), hemolytic uremic syndrome (HUS), and renal failure (Joseph et al., 2020; Pakbin et al., 2021). HUS occurs in ∼10% of patients (Allerberger et al., 1997; Nataro and Kaper, 1998; Karch et al., 2005). A well-controlled expression of these virulence factors determines the success of EHEC in colonizing the recto-anal mucosa of ruminants and colonic mucosa of humans to cause disease.
The Locus of Enterocyte Effacement
The locus of enterocyte effacement (LEE) was first detected in the EPEC strain (McDaniel et al., 1995) and thereafter in EHEC strains (Kaper et al., 2004). LEE is a chromosomally encoded pathogenicity island (PAI) that harbors genes responsible for a type III secretion system (T3SS) including intimin (Eae), its translocated intimin receptor (Tir), LEE-encoded regulator (Ler), chaperones, and many other effector proteins (Kaper et al., 1999; Robins-Browne and Hartland, 2002). The E. coli secreted protein A (EspA) is a translocator protein that forms a long filamentous extension through which effector proteins are delivered to the host cell (Garmendia et al., 2005). The EspB and EspD translocator proteins form a translocation pore in the plasma membrane of the host cell (Abe et al., 1997; Kresse et al., 1999; Buttner and Bonas, 2002; Deng et al., 2004; Garmendia et al., 2005).
Intimin binds to Tir to establish the intimate contact required for colonization (Dean-Nystrom et al., 1998). The synthesis of LEE-encoded, and non-LEE-encoded, effectors and their translocation into a host cell are tightly regulated in response to different signals. The expression of T3SS of EHEC allows colonization of ruminants (Naylor et al., 2003; Low et al., 2005), whereas deletion of genes in T3SS reduces colonization (Dziva et al., 2004; Naylor et al., 2005).
However, how LEE-encoded and non-LEE-encoded virulence factors converge to achieve T3SS is unclear.
Thus, well-defined understanding of LEE-encoded and non-LEE-encoded effectors responsible for T3SS and regulation mechanisms that control their expression at transcriptional and post-transcriptional levels in different compartments of the gastrointestinal tracts of humans and ruminants is important to design effective tools to prevent EHEC colonization of humans and ruminants. Detailed understanding of the interactions among different virulence factors to achieve AE lesions and associated HC and HUS in humans will help to identify crucial virulence factors that can be targeted for intervention.
Transcriptional Regulation of LEE Expression
LEE is ≈35 kb comprising 41 reading frames that are structured into 5 distinct polycistronic operons designated as LEE1 to LEE5 (Figure 1) (Elliott et al., 2000). The success of colonization and survival of EHEC in its host depends on its ability to respond appropriately to host environmental cues by coordinating and regulating the transcription of its virulence genes. As a result, LEE-encoded and non-LEE-encoded virulence factors are under tight regulation at both at transcriptional and post-transcriptional levels involving numerous direct and indirect regulatory pathways that depend on various environmental conditions (Mellies and Barron, 2006; Mellies et al., 2007; Franzin and Sircili, 2015).

Genetic organization of LEE encoding T3SS and T3SS2. Green arrows showed expression induction pathways, whereas red blunted lines showed downregulation of expression. AHL, acyl-homoserine lactone; AI-3, autoinducer 3; ArgR, arginine transcription regulator; CM, cell membrane; CpxA, sensor kinase; CpxR, response regulator; Cra, transcription factor; CutR, cysteine-responsive regulator; EA, ethanolamine; EivF, Escherichia coli type three secretion system regulator F; Epi, epinephrine; EtrA, E. coli type three secretion system 2 regulator A; ExuR, transcription factor; FadR, transcription regulator; FusR, fucose response regulator; Hha, hemolysin expression-modulating protein; H-NS, histone-like nucleoid-structuring; kdpE, response regulator protein E; LEE1–LEE5, locus of enterocyte effacement 1–5; Ler, LEE-encoded regulator, eutR, ethanolamine utilization regulator gene; LrP, leucine response regulatory protein; FusK, fucose-sensing kinase; NAG, N-acetylglucosamine; NagC, N-acetylglucosamine-6 phosphate sensing protein; NE, norepinephrine; pchA, perC homologue A gene; pchB, PerC homologue B gene; PPGPP, guanosine tetraphosphate; QseA, quorum-sensing E. coli sensor regular A; QseB, quorum-sensing E. coli response regular B; QseC, quorum-sensing E. coli sensor C; QseE, quorum-sensing E. coli sensor E; QseF, quorum-sensing E. coli response regular F; RcsB, response regulator; SspA, stringent starvation protein A; T3SS2, type III secretion system 2.
LEE-Encoded Regulators of LEE
Ler is an LEE1 operon–encoded transcriptional regulator of LEE expression in EHEC. It functions as the main receiver of several regulatory proteins, thereby controlling AE phenotype (Mellies et al., 2007). Since the expression of Ler is a key to virulence gene regulation, its expression is important for the pathogenesis of EHEC infection (Elliott et al., 2000). Thus, Ler expression is regulated by several regulatory factors, some of which are encoded within the LEE region, whereas others are outside the LEE region (Figures 1 and 2) (Franzin and Sircili, 2015; Turner et al., 2019). Previous study (Elliott et al., 2000) showed that mutation of ler gene in an EHEC strain disrupted the ability to form AE lesions on human intestinal epithelial cells, suggesting that the protein is essential for virulence expression.
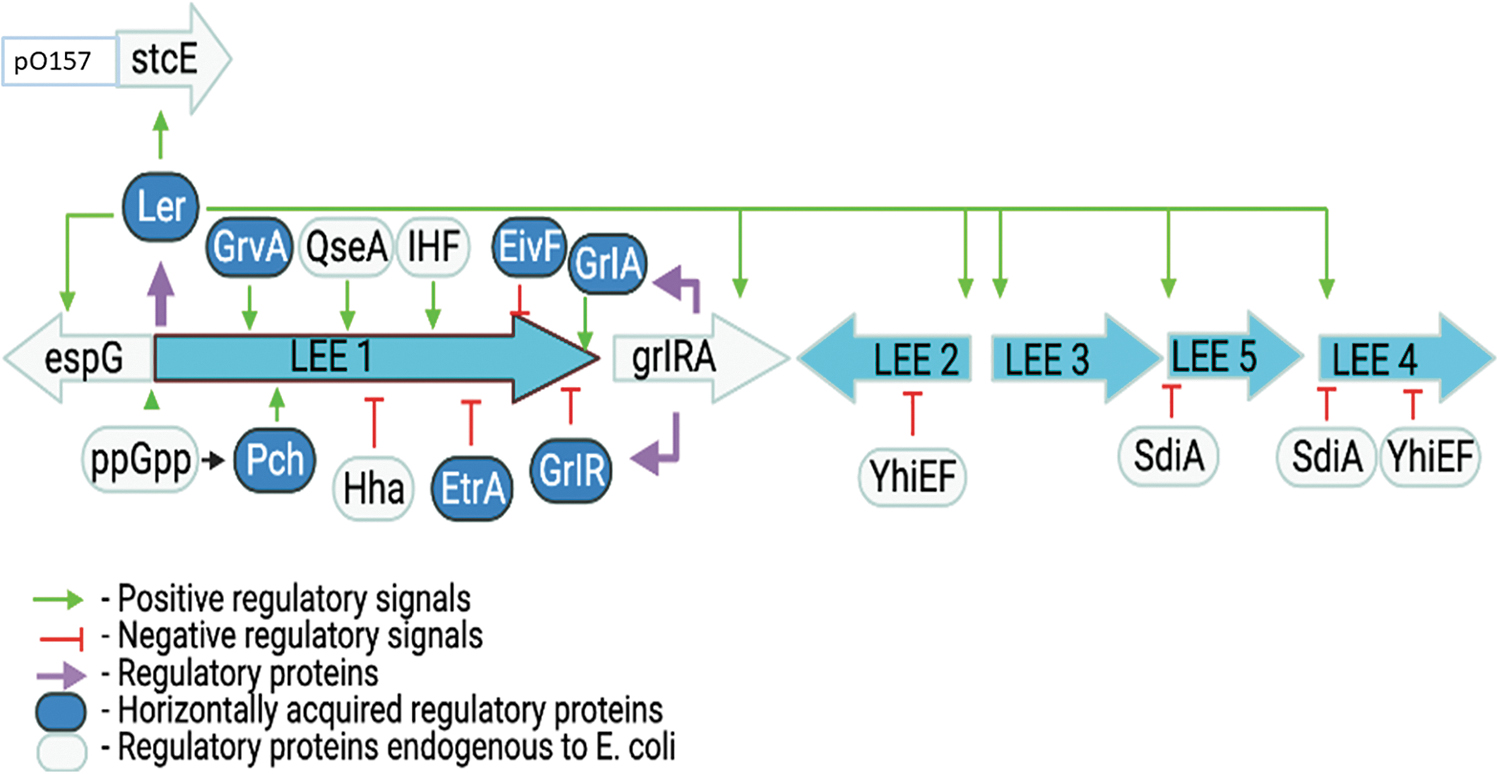
Ler-mediated regulation of the LEE pathogenicity island and LEE-encoded genes in EHEC; adapted from Mellies et al. (2007) with permission. EHEC, enterohemorrhagic Escherichia coli; EivF, E. coli type III secretion system 2 regulator F; espG, E. coli secreted protein G gene; EtrA, E. coli type III secretion system 2 regulator A; GlrR, global regulator of LEE repressor; GrlA, global regulator of LEE activator; grlRA, global regulator of LEE repressor and activator genes; GrvA, global regulator of virulence activator; Hha, hemolysin expression-modulating protein; IHF, integration host factor; LEE, locus of enterocyte effacement; Ler, LEE-encoded regulator; Pch, plasmid-encoded regulator C homolog; ppGpp, guanosine tetraphosphate; QseA, quorum-sensing E. coli response regular A; SdiA, suppressor of division inhibitor A; stcE, encodes a zinc metalloprotease that cleaves the C1 esterase inhibitor.
Ler regulates transcription of LEE2, LEE3, LEE4, and LEE5 operons (Figs. 1 and 2) (Elliott et al., 2000). In addition, Ler regulates the non-LEE stcE gene that encodes a metalloprotease (Elliott et al., 2000; Lathem et al., 2002; Li et al., 2004; Grys et al., 2005; Hews et al., 2017) that facilitates intimate adherence of EHEC to the host cell (Grys et al., 2005). Mutation of ler blocks the expression of T3SS proteins, whereas complementation restores it, suggesting that Ler regulates all vital genes essential to form AE lesions; thus, Ler protein is a global regulator of virulence genes in EHECs LEE (Figs. 1 and 2) (Elliott et al., 2000).
Two important LEE-encoded regulators, a global regulator of LEE activator (GrlA) and a global regulator of LEE repressor (GlrR), were reported in human EHEC and EPEC, and mouse Citrobacter rodentium strains (Deng et al., 2004). GrlA is responsible for the positive regulation of ler in the expression of LEE genes. Mutation of grlA gene markedly reduced the expression of LEE1, LEE2, and LEE5, whereas its complementation restored it to baseline level (Deng et al., 2004), suggesting that GrlA activates LEE gene transcription. On the contrary, the expression of LEE1 increased in the grlR mutants, suggesting that GrlR represses transcription of the LEE genes (Figure 2).
Another study (Barba et al., 2005) showed that GrlA attaches to the LEE1 promoter and positively regulates Ler synthesis. In this experiment, they also showed that the synthesis of the GrlA operon is under the control of Ler, establishing a positive regulatory loop so that the proper level of Ler protein is synthesized to activate the expression of the LEE genes. Using a standard yeast two-hybrid system, another study in EPEC demonstrated that GrlR specifically binds to GrlA (Creasey et al., 2003). The binding of GrlR to GrlA suppresses LEE gene expression by inhibiting GrlA interaction with the LEE1 promoter (Huang and Syu, 2008; Padavannil et al., 2013) (Figure 2).
Non-LEE-Encoded Regulators of LEE
Several transcriptional regulatory proteins indirectly control EHECs AE lesion formation via the synthesis of Ler (Mellies et al., 2007). The regulatory protein, histone-like nucleoid-structuring (H-NS) (Bustamante et al., 2001; Umanski et al., 2002; Haack et al., 2003), downregulates the expression of LEE and other horizontally acquired mobile genes characterized by lower GC or higher AT contents than the resident genome across Enterobacteriaceae (Navarre et al., 2007). The H-NS is repressor of EPEC virulence genes (Bustamante et al., 2001), and Ler acts as an antirepressor that overcomes the H-NS-mediated silencing on the LEE2/LEE3 divergent promoter region (Table 1).
Summary of Major Locus of Enterocyte Effacement- and Non-Locus of Enterocyte Effacement-Encoded Transcriptional Regulators of Genes on Locus of Enterocyte Effacement
BipA, bactericidal/permeability increasing (BPI)-inducible protein A; Cpx, envelope stress response regulators of a two-component system (TCS) consisting of a sensor histidine kinase (CpxA) and a cytoplasmic response regulator (CpxR); EHEC, enterohemorrhagic Escherichia coli; EivF, E. coli type III secretion system 2 regulator F; EtrA, E. coli type III secretion system 2 regulator A; EutR, ethanolamine utilization (eut) regulator; FusK/R, proteins involved in the bacterial two-component regulatory system, FusK is fucose sensing kinase, whereas FusR is fucose response regulator; GadE, a master regulator of glutamate-dependent acid resistance; GlrR, global regulator of LEE repressor; GrlA, global regulator of LEE activator; GrvA, global regular of virulence A; Hha, hemolysin expression-modulating protein; H-NS, histone-like nucleoid-structuring protein; LEE, locus of enterocyte effacement; Ler, LEE-encoded regulator; LeuO, regulator of leucine biosynthesis operon; PchA, plasmid-encoded regulator C homologue A; PchB & PchC, plasmid-encoded regulator C homologue B and C; Per, plasmid-encoded regulator; RgdR, EHEC regulator; RpoN, RNA polymerase nitrogen-limitation; SdiA, suppressor of division inhibitor; SspA, stringent starvation protein A.
Another study (Wan et al., 2016) demonstrated the upregulation of several virulence genes on plasmid pO157 in EHEC strain with h-ns deletion mutant compared with the wild-type strain. The authors showed that the h-ns mutant EHEC strain showed a greater ability to colonize mice guts. Mice infected with the isogenic mutant clone had a significantly shorter survival rate than those infected with the wild-type strain. RNA-Seq analysis showed that many virulence genes, including those encoding T3SS, were upregulated in h-ns mutant suggesting that H-NS downregulates the expression of LEE virulence genes (Table 1).
Similarly, hemolysin expression-modulating protein (Hha) was shown to downregulate Ler expression, which causes reduced expression of LEE4 (Sharma and Zuerner, 2004). These authors (Sharma and Zuerner, 2004) showed that inactivation of the hha gene in EHEC increased the synthesis of Ler and enhanced adherence to HEp-2 cells in culture. They also showed that complementing the mutant with hha-containing plasmid restored the synthesis to the level expressed in the wild-type EHEC strain.
In EHEC, LEE expression can be induced by other proteins such as integration host factor (Friedberg et al., 1999), factor for inversion stimulation (Goldberg et al., 2001), bactericidal permeability-increasing protein (Grant et al., 2003), and RgdR (Flockhart et al., 2012) (Table 1).
Genome sequencing of E. coli O157:H7 uncovered a gene cluster encoding components of a second cryptic T3SS (T3SS2) (Fig. 1). Deleting E. coli type III secretion system 2 regulator A (etrA) and E. coli type III secretion system 2 regulator F (eivF) in E. coli O157:H7 considerably increased LEE-encoded proteins expressions, thereby enhancing adhesion to human intestinal cells. The EtrA and EivF negatively regulate the transcription of genes within LEE (Zhang et al., 2004), whereas EtrB upregulates the expression of LEE by repressing the transcription of etrA and eivF genes (Luzader et al., 2016).
Plasmid-encoded regulator (Per)-like proteins that regulate the expression of LEE in EHEC were reported by two independent research groups (Iyoda and Watanabe, 2004; Porter et al., 2005). Iyoda and Watanabe (2004) compared the gene sequence of Per-like proteins in EHEC with EPEC Per proteins and found that the Per-like molecules in EHEC have similarities to the perC of EPEC. In the EHEC, these proteins are prophage-encoded. They are named plasmid-encoded regulator C homologs PchA, PchB, and PchC. They regulate ler expression with PerC in EPEC (Iyoda and Watanabe, 2004; Fukui et al., 2016). On the contrary, another study concluded that the EHEC PchA protein directly activates the LEE1 promoter, and then, Ler activates LEE2, LEE3, LEE4, and LEE5 promoters (Porter et al., 2005). The same authors (Porter et al., 2005) reported that PchB and PchC are inactive and do not play any role in activating the expression of LEE1.
The CpxAR envelope stress response system of EHEC plays an important role in the regulation of LEE gene expression. The Cpx is regulated by sensor histidine kinase (CpxA) and a cytoplasmic response regulator (CpxR) (Hung et al., 2001). A study demonstrated that mutation in cpxA gene disrupted adherence of EHEC to HeLa cells (De la Cruz et al., 2016). Compared with the wild-type strain, the isogenic cpxA mutant EHEC strain was unable to kill the larvae of Galleria mellonella, a eukaryotic model to determine pathogenicity or virulence of Salmonella, EPEC, and other enteric and non-enteric pathogens (O'Loughlin et al., 2010; Debnath et al., 2013; Bontemps-Gallo et al., 2015; De la Cruz et al., 2015; Thomassin et al., 2015) for their hosts. Thus, CpxA indirectly prevents EHECs AE phenotypes by activating the expression of CpxR, a protein that represses GrlA and Ler transcription.
RNA polymerase nitrogen-limitation (RpoN) is one of the multiple RNA polymerase sigma subunits needed to regulate stress resistance and virulence genes in E. coli. Inactivation of rpoN in EHEC strain led to significant downregulation of genes within the five LEE operons, including grlA (Riordan et al., 2010). During exponential growth, RpoN (σN or sigma 54) and its cognate nitrogen regulatory protein C (NtrC-σN) initiate transcription of motility regulator flhD (Mitra et al., 2012; Riordan and Mitra, 2017).
The FlhD activates transcription of σS antagonist, FliZ. The expression of FliZ represses σS activity, leading to the downregulation of glutamate-dependent acid resistance (GDAR) central regulator gadE and concomitant upregulation of the LEE (Mitra et al., 2012; Riordan and Mitra, 2017). The GDAR-dependent repression of σS by σN/FliZ is through the GadX-GadE pathway (Mitra et al., 2014). The σS represses LEE expression through downregulation of the PchA-Ler activation pathway and downregulation of gadE since GadE is known to repress LEE transcription directly or indirectly through GadX (Iyoda and Watanabe, 2005; Branchu et al., 2014).
Two studies (Tomoyasu et al., 2005; Iyoda and Watanabe, 2005) identified a DNA fragment containing clpX and clpP genes whose product positively regulates the expression of LEE in E. coli O157. Both groups demonstrated that deletion of the clpXP gene significantly reduced the expression of virulence factors secreted by T3SS and downregulated transcription from all five LEE operons. Inactivation of rpoS, the gene encoding a sigma factor that is a substrate for ClpXP protease, partly restored the transcription from all the LEE promoters and the expression of virulence proteins in clpPX mutant. It has been observed that deletion of rpoS triggers transcription of pchA, one of the positive regulators for LEE genes in EHEC (Iyoda and Watanabe, 2005).
Deleting grlR, a negative regulatory gene within LEE, significantly enhanced LEE expression even in the clpXP mutant, signifying that the GrlR level might be controlled by ClpXP, which is essential for the ClpXP-dependent expression of LEE in EHEC. These studies indicate that ClpXP protein positively regulates LEE expression, and the regulation happens through the control of RpoS and GrlR levels in EHEC. In addition, two independent groups of researchers (Laaberki et al., 2006; Vijayakumar et al., 2004) have shown that RpoS positively regulate the LEE3 operon and tir in a specific EHEC strain. However, it should be noted that RpoS regulation of LEE expression differs with EHEC strain and environmental conditions (Laaberki et al., 2006).
In addition to well-characterized LEE-encoded and non-LEE-encoded regulators that control AE phenotype, a total of 10 fimbrial genes and 13 non-fimbrial adhesins were found in EHEC (Hayashi et al., 2001; Perna et al., 2001). Of which, the long polar fimbriae (Jordan et al., 2004; Farfan et al., 2011; Lloyd et al., 2012), E. coli common plus (Rendon et al., 2007), the autotransporter protein, EhaG (Totsika et al., 2012), and the pO157 virulence plasmid-encoded zinc metalloprotease (StcE) (Lathem et al., 2004; Grys et al., 2005) are involved in EHEC adhesion to epithelial cells and host colonization Figures 1 and 2).
Post-Transcriptional Regulation of LEE Expression
Most LEE virulence gene regulation occurs at the transcription level; however, post-transcriptional and post-translational control of LEE gene expression also happens in EHEC (Franzin and Sircili, 2015). The DsrA is a non-coding RNA that regulates transcription of LEE by antagonizing transcriptional silencing caused by H-NS protein (Gottesman, 1995; Lease et al., 1998) and activating translation of a conserved alternative sigma factor (Lease et al., 1998; Majdalani et al., 1998; Laaberki et al., 2006).
Small RNAs (sRNA) post-transcriptionally control several processes in bacteria. Base pairing of sRNAs near ribosome-binding sites in mRNAs blocks translation, mostly involving the RNA chaperone, Hfq (Hoekzema et al., 2019). In EHEC, Hfq affects LEE expression by interacting with ler mRNA (Hansen and Kaper, 2009; Shakhnovich et al., 2009; Kendall et al., 2011). Hfq acts as a negative regulator of most LEE genes involved in T3SS and AE phenotypes in EHEC EDL933 strain (Majdalani et al., 1998), while it also acts as a positive regulator of LEE genes in EHEC 86-24 strain (Kendall et al., 2011). Deleting hfq gene also reduced the expression of a two-component system qseBC, which activates Shiga toxin–encoding genes (Table 1 and Figure 2).
Quorum Sensing–Mediated Regulation of LEE
Chemical signals and metabolites produced by the host and gut microbiota control LEE expression in different parts of the gastrointestinal tract (Hughes et al., 2010; Sharma et al., 2022). Mammalian host releases epinephrine (Epi) and norepinephrine (NE) hormones during stress. EHECs quorum sensing (QS) E. coli regulator C (QseC) binds these host hormones or bacterial autoinducer (AI-3). Sensing of Epi and NE is achieved through two histidine kinase response regulator systems, QseBC and QseEF, which in turn activate QseA, which activates transcription of ler, by binding to the P1 and P2 promoter region (Sperandio et al., 2002; Reading et al., 2007, 2009; Njoroge and Sperandio, 2012) (Figure 3, Table 2).
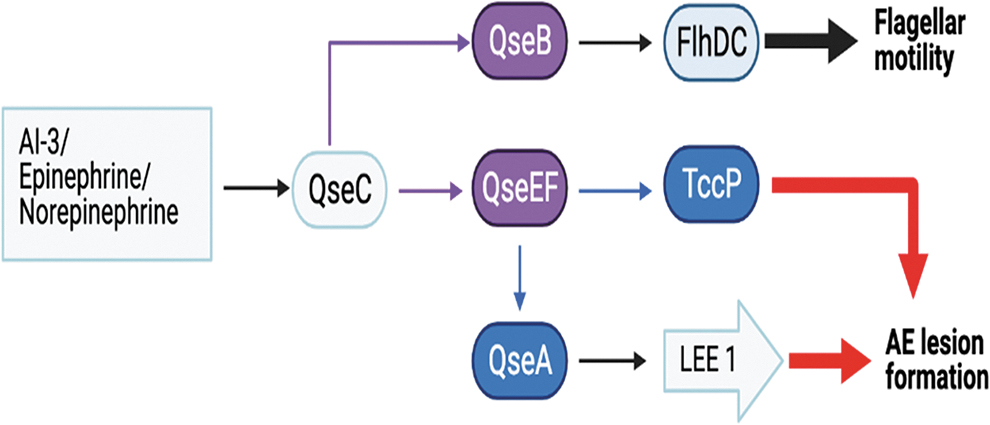
Quorum sensing–mediated regulation of flagellar motility and attaching and effacing lesion formation in EHEC; adapted from Mellies et al. (2007) with permission. AI-3, autoinducer 3; EHEC, enterohemorrhagic Escherichia coli; FlhDC, the heterohexameric operon that encodes the transcription factors FlhD and FlhC that induced transcription of flagellar apparatus, motility, and chemotaxis; QseA, quorum-sensing E. coli regulator A; QseB, quorum-sensing E. coli response regulator B; QseC, quorum-sensing E. coli sensor C; QseEF, in the two-component regulatory system, quorum-sensing E. coli sensor E (QseE) and quorum-sensing E. coli response regulator F; TccP, Tir cytoskeleton coupling protein.
Summary of Quorum Sensing Regulators of Locus of Enterocyte Effacement Genes in Enterohemorrhagic Escherichia coli
KdpE, a response regulator that activates the transcription of homeostasis genes in response to salt-induced osmolarity and virulence genes in response to changes in metabolite concentrations; LEE, locus of enterocyte effacement; QseA, quorum-sensing E. coli regulator A; QseC, quorum-sensing E. coli sensor C; QseD, quorum-sensing E. coli response regulator D.
The QseC phosphorylates response regulator, QseB, which activates flhDC and fliA genes that encodes flagellar biosynthesis (flhDC) and motility (fliA) (Figure 3 and Table 2) (Sperandio, 2002; Sperandio et al., 2002; Clarke and Sperandio, 2005; Clarke et al., 2006; Hughes et al., 2009; Meng et al., 2016). The sensor kinase QseE responds to sulfate, phosphate, and host-derived Epi to phosphorylate its cognate response regulator QseF, which ultimately stimulates ler expression (Reading et al., 2010). The QseEF also controls the synthesis of EspFu through AI-3, Epi, and NE signaling (Reading et al., 2007). The sensor kinase QseC binds Epi or NE or EHEC produced AI-3 and phosphorylates KdpE, which induces increased ler expression (Hughes et al., 2009) (Table 2 and Figure 4).
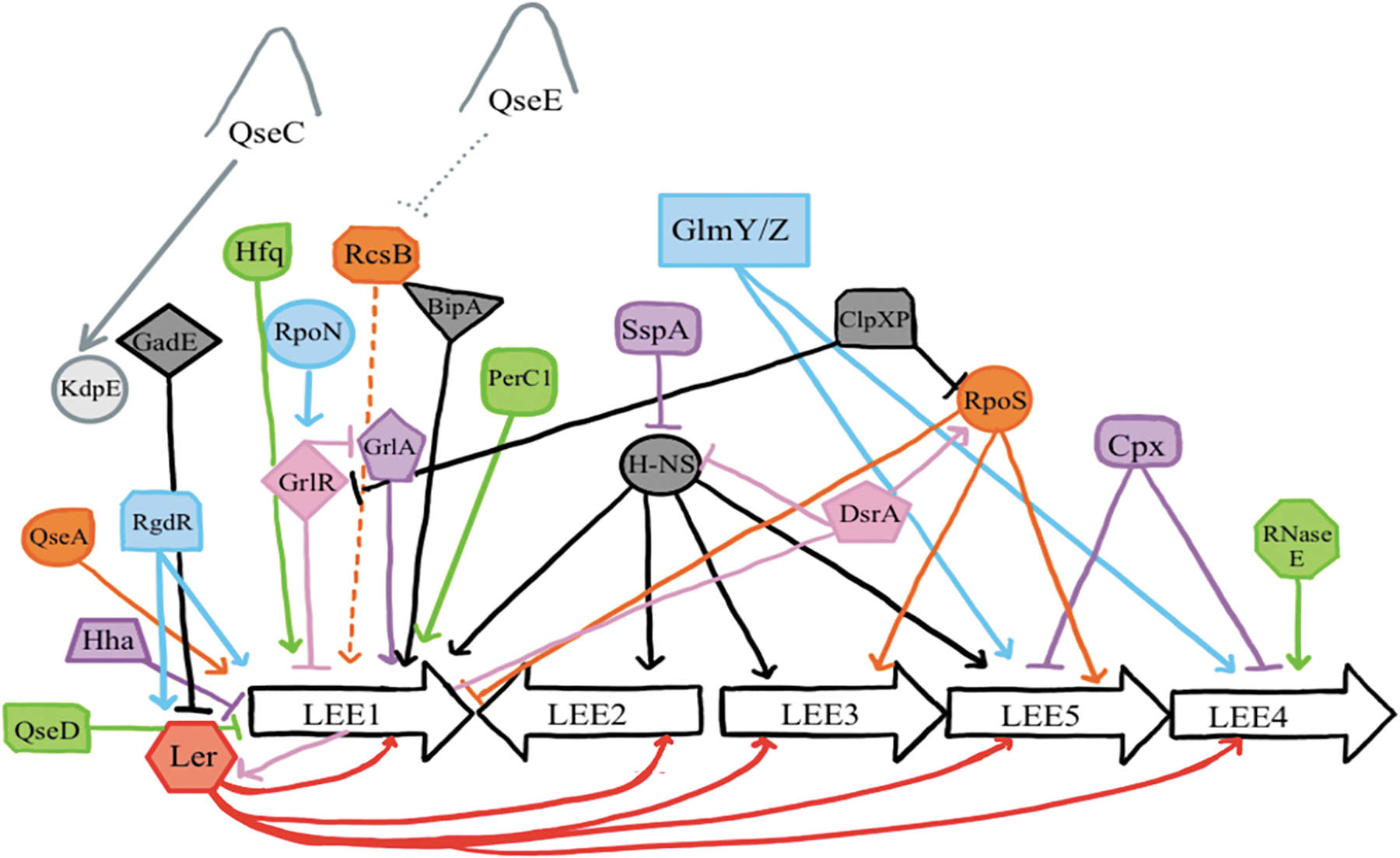
Diagrammatic illustration of a complex network of LEE genes regulation in EHEC. Pointed arrows represent activation (positive regulation), whereas the blunt arrows show repression. The figure was adapted from Franzin and Sircili (2015) with permission. σS (RpoS), RNA polymerase sigma S; σE (RpoE), RNA polymerase E; BipA, bactericidal/permeability-increasing (BPI)-inducible protein A; ClpXP, a two-component protease composed of ClpX, an ATP-dependent chaperone that recognizes and unfolds specific substrates, and ClpP, a serine protease; DsrA, non-coding RNA that regulates both transcription and translation; EHEC, enterohemorrhagic Escherichia coli; LEE, locus of enterocyte effacement; RpoS, RNA polymerase S or a stress sigma factor; GadE, a master regulator of glutamate-dependent acid resistance; Glm Y/Z, small RNAs that regulate transcription of glmS gene that encodes glutamine-fructose-6-phosphate transferase; Glm Y- activate transcription of glmS and Glm Z stabilizes a glmS transcript; GrlA, global regulator of LEE activator; GlrR, global regulator of LEE repressor; Hfq, small RNA chaperone; Hha, hemolysin expression-modulating protein; H-NS, histone-like nucleoid-structuring protein; KdpE, a response regulator protein E; Ler, LEE encoded regulator; perC1, plasmid-encoded regulator C1; QseA, quorum-sensing E. coli response regulator A; QseC, quorum-sensing E. coli sensor C; QseD, quorum-sensing E. coli sensor D; QseE, quorum-sensing E. coli sensor E; RcsB, response regulator B; RgdR, regulator protein; RNase E, RNA digesting enzyme E; RpoN, RNA polymerase nitrogen-limitation; SspA, stringent starvation protein A.
QseC also phosphorylates response regulators, KdpE, and QseF. The KdpE activates transcription of the genes in LEE operons. The QseF and QseB together activate the transcription of sRNA glmY, which post-transcriptionally further regulate the expression of the LEE and espFU (Njoroge et al., 2013). Deleting qseC in rabbit EPEC model severely attenuated virulence (Clarke et al., 2006; Rasko et al., 2008), and a qseF deletion mutant of EHEC does not form AE lesions (Reading et al., 2007). The non-LEE-encoded A (nleA) expression is ler-dependent and in part induced by the Global response to DNA damage induced stress (SOS) response, while expression of espFU is controlled by the QseA and QseEF pathways.
Whole-genome transcriptional analysis of E. coli O157:H7 demonstrated that an optimal level of NE in the gut serves as an important signal for the bacteria to upregulate virulence gene expressions that enhance colonization of the reservoir animals and disease in susceptible hosts (Sharma et al., 2022). In conclusion, QS is one of the main pathways that activate ler expression. QS can be an ideal target for novel therapeutic and prevention approaches against EHEC colonization and infection.
LEE Expression (On/Off) Mechanisms at Different Parts of Host Gastrointestinal Tract
EHEC uses its acid resistance (AR) mechanisms to avoid death from the low pH of the rumen in cattle and stomach in humans. These include (1) glucose repressed or oxidative, (2) glutamate-dependent, and (3) arginine-dependent mechanisms (Foster, 2004; Price et al., 2004; Richard and Foster, 2004). In the rumen, EHEC uses the SdiA protein, which is a 240-aminoacid protein within the LuxR family of transcriptional regulators, to detect acyl-homoserine lactones (AHLs) secreted by other groups of bacteria (Hughes et al., 2010) to activate the expression of the GDAR system (Figures 1 and 4).
Inactivation of sdiA eliminates the ability of EHEC to sense AHL and control LEE and gad gene expression (Sheng et al., 2013). The authors showed that the sdiA mutant failed to grow well inside the rumen compared with the wild type as the former could not resist the host's low pH ruminal environment due to accumulation of anion from volatile fatty acids produced in the rumen by normal microbiota. According to this study, SdiA directly represses the expression of ler, while it indirectly drives gad expression. The resident microbiota produces AHL that binds to SdiA, which consequently became activated to induce synthesis of GadX that triggers transcription of glutamate decarboxylases A, B, and C (gadA, gadB, and gadC) genes resulting in glutamate decarboxylases A, B, and C (GadA, GadB, GadC) proteins.
The GadA and GadB convert glutamate to γ-aminobutyric acid (GABA) and CO2 (Lin et al., 1996; Castanie-Cornet et al., 1999; De Biase et al., 1999). The GadC transports glutamate into the bacterium while removing GABA from the bacterium. The pH homeostasis is maintained by displacement of the α-carboxyl group of amino acids with a proton transported from the environment into the bacterium's cytoplasm. Cytoplasmic glutamate is restored by antiporter GadC. The AHL-SdiA pathway downregulates ler expression (Hughes et al., 2010) while upregulating the expression of the Gad system for AR in the rumen.
In the human stomach, glutamate-dependent mechanisms regulate AR (Umanski et al., 2002; Ma et al., 2003; Tucker et al., 2003; Hommais et al., 2004). GadE protects EHEC by stimulating the glutamate-dependent AR system and blocks the needless expression of the LEE genes when EHEC passes through the stomach (Vanaja et al., 2009). The authors showed that inactivation of gadE causes a marked increase in the expression of ler and other multiple genes encoded in LEE. They also showed that disruption of ler in the gadE mutant strain overturned the impact of gadE deletion on LEE expression, indicating that Ler is required for LEE repression by GadE.
The expression of ler, espA, and tir increased up to twofold in gadE mutant strains compared with the wild type (Tree et al., 2011). Complementation of the mutant with the gadE gene restored the effect to the level in the wild type. The Gad system is regulated by several environmental factors and global regulatory proteins such as H-NS, catabolite repressor protein, and alternate sigma factors (RpoS and RpoN) (Lin et al., 1996; Castanie-Cornet et al., 1999; De Biase et al., 1999; Tramonti et al., 2002; Mitra et al., 2012).
In the small intestine, the high glucose content and glycolysis downregulate the ler transcriptional regulators KdpE and Cra (Njoroge et al., 2012), thereby downregulating ler expression. Similarly, fucose, a component of cell surface glycans, increases in the mammalian intestine by cleavage from mucin glycoproteins by fucosidases produced by commensal bacteria Bacteroides thetaiotaomicron (Snider et al., 2009). Fucose binds sensor histidine kinase fusK, which phosphorylates response regulator FusR that downregulates ler expression (Figure 1 and Table 1) (Pacheco et al., 2012).
In the large intestine, gluconeogenic conditions of the large intestine activate the KdpE and Cra ler activation, which directly induce LEE transcription (Nakanishi et al., 2009; Njoroge et al., 2013). Ethanolamine (EA), a bacterial and animal cell membrane component released into the gut lumen upon normal epithelial cell turnover, increases the expression of QS regulators QseA, QseC, and QseE, and ler as well as expression of LEE operons 1–5 (Kendall et al., 2012). The QseE controls LEE expression by inhibiting the positive-acting RcsB response regulator (Njoroge and Sperandio, 2012). This regulation occurs through the proteins KdpE, a response regulator that also senses osmotic stress, and Cra, both of which bind to ler regulatory DNA. Deletion of KdpE and Cra resulted in the loss of AE lesion formation by EHEC in vitro (Njoroge et al., 2012) (Figure 1 and Table 2).
The EA utilization gene (eut) is necessary for EA utilization as a nitrogen source, conferring EHEC growth during the stationary phase (Bertin et al., 2011) in the bovine and human large intestine and signaling for expression of the T3SS (Bertin et al., 2011). Nutrient deprivation activates the stringent response that induces the production of ppGpp, which promotes the expression of the LEE transcriptional activators pchA, pchB, and nleA. Thus, the synthesis of PchA/B induces the expression of ler (Nakanishi et al., 2006; Schwidder et al., 2011). The nleA can also be activated through QseE and transcription activator RcsB (Figures 1 and 4).
The E. coli stringent starvation protein A (SspA) is needed for transcriptional activation of certain horizontally acquired genes in E. coli and aids in its survival during nutrient deprivation and protracted stationary phase (Williams et al., 1991). In addition to this role, Hansen and Jin (2012) showed that SspA activates the expression of EHEC virulence genes by reducing the level of H-NS within the bacterial cell. The authors also demonstrated that the inactivation of sspA genes in EHEC reduced the expression of LEE virulence genes, and the mutant strain was rendered deficient at forming AE lesions on HEp-2 cells. Enhanced ler expression alleviates the expression of LEE in an sspA mutant, indicating that the level of Ler in the mutant is inadequate to counter H-NS-mediated repression as the level of the H-NS is two times greater in an sspA mutant compared with the wild type.
Butyrate, a short-chain fatty acid found within the intestine, increases the transcription of LEE genes in EHEC, leading to enhanced bacterial adherence (Franzin and Sircili, 2015). Another study showed that the expression of the regulator of leucine biosynthesis operon (leuO), regulatory protein, increased by butyrate through leucine responsive regulatory protein, which is also required for butyrate-induced responses of LEE genes (Takao et al., 2014) (Figures 1 and 4 and Table 1).
Regulation of Shiga Toxin Expression
The cause of epidemic dysentery, S. dysenteriae type 1 was first discovered by Shiga (1889). The verocytotoxin was discovered in 1977 (Konowalchuk et al., 1977) and later shown to be biologically and structurally similar to the Shiga toxin of S. dysenteriae type 1, and named Shiga-like toxin (O'Brien et al., 1982; O'Brien and Laveck, 1983). EHEC produces two main kinds of Stxs, Stx1 and Stx2 (Scotland et al., 1985).
Stxs are a group of bacterial AB5 protein toxins that inhibit protein synthesis in sensitive eukaryotic cells. Stx removes an adenine from the 28S rRNA of the 60S ribosome and prevents protein synthesis. The A subunit achieves the N-glycosidase or enzymatic function of the toxin, whereas the B subunit binds to the cell surface receptor globotriaosylceramide (GB3) (Thorpe, 2004). The A and B subunits of the Sxts were encoded by stxA and stxB genes, respectively (Griffin and Tauxe, 1991). EHEC stx1 and stx2 genes are encoded by separate prophages integrated into the chromosome. Both toxins are produced during the phage lytic cycle activated by various environmental cues (Perna et al., 2001).
Environmental signals such as levels of iron, growth phase, antibiotics, reactive oxygen species, temperature, and QS regulate the expression of Stx (Pacheco and Sperandio, 2012). The fatality attributed to EHEC infections is mainly due to production and secretion of a potent Stx (O'Brien et al., 1984). Stx causes apoptosis of endothelial cells in the intestine and the urinary tract, inhibiting protein synthesis via inhibition of aminoacyl-tRNA binding (Brown et al., 1986).
EHEC regulates expression of its virulence genes in response to several host and microbial-derived signals in the gastrointestinal tract (Mellies and Lorenzen, 2014; Connolly et al., 2015; Bäumler and Sperandio, 2016; Turner et al., 2019). A study (O'Brien et al., 1983) demonstrated that low-iron environments lead to activation of stx 1AB, suggesting that the level of iron affects the production of Stx1. High iron levels repress the activity of the stx 1AB iron-dependent promoter, and specifically, stx1 expression is derived through the iron-responsive Fur repressor. However, iron does not appear to affect the expression of stx 2AB genes (Calderwood and Mekalanos, 1987).
The level of Stx genes expression depends on bacterial growth phase. It was shown (Bergholz et al., 2007) that Stx gene expression were markedly upregulated during the stationary phase compared with the exponential phase. Similarly, another study (Konowalchuk et al., 1978) also suggested that Stx production increases during stationary phase, and maximum cytotoxic activity in EHEC culture was observed during 24 h after incubation. The kinetic patterns for the release of the toxins from Shiga toxin–producing E. coli (STEC) strains showed that both Stx1 and Stx2 were released into the growth medium during exponential growth phase (Yokoyama et al., 2000). A rpoS-deficient mutation did not alter the levels of extracellular Stx1 and Stx2, supporting the idea that the toxins are produced during the exponential growth phase (Yokoyama et al., 2000).
Antibiotics enhance the production of Stxs due to their effect on the induction of the phage lytic cycle (Zhang et al., 2000; McGannon et al., 2010). As a result, antibiotics, especially those that interfere with DNA replication such as quinolones, are contentious for treating infections caused by STEC (Zhang et al., 2000). It was shown that while antibiotics that affect DNA synthesis prompt Stx production, other antibiotics which act on other cellular processes, such as cell wall synthesis and protein synthesis, impede Stx production (Wong et al., 2000).
Host cells such as neutrophils and cell products, including hydrogen peroxide (H2O2) might cause bacterial DNA damage and potentially activate an SOS response, bacterial response to DNA damage (Imlay and Linn, 1987; Lobysheva et al., 1999). EHEC exposure to H2O2 in vitro led to upregulation of Stx production and increased phage titers (Wagner et al., 2001). Similarly, another study (Los et al., 2010) reported that H2O2 induces Stx production at a level similar to the amount produced by norfloxacin, a quinolone antibiotic. The same authors also observed increased Stx production after incubating EHEC with human neutrophils, suggesting that neutrophil-derived products can trigger an SOS response resulting in Stx release. In addition, to finding ways to block Stx production, further investigation on effective alternative therapeutic and preventive approaches, including monoclonal antibodies, toxin receptor analogs, and vaccination, are of significant importance.
Conclusions
EHEC uses two major virulence factors, the LEE PAI and phage-encoded Shiga toxin, to drive pathogeneses in the susceptible host. Well-controlled expression of these virulence factors is key to the host's successful colonization, which may lead to HC and HUC. EHEC regulates the expression of its virulence factors such as Stx, LEE-encoded cocktails of effectors that are delivered to host cells through sophisticated T3SS, which cause AE lesion, and non-LEE-encoded effectors depending on multiple signals derived from the host, microbiota, nutrients and oxygen tension, and other factors in the microenvironmental niche. Thus, a thorough understanding of the mechanism and proteins involved in the regulation of virulence genes is crucially important to develop preventive and control measures for this important pathogen.
Footnotes
Acknowledgments
We thank all our Dairy Health Research Group members in Dr. Kerro Dego's Laboratory at the Department of Animal Science, The University of Tennessee, Knoxville, for reading and editing the article.
Authors' Contributions
B.D.G.: Conceptualization (lead); writing—original draft (lead). S.B.: Writing—original draft (supporting). H.C.: Writing—original draft (supporting). G.E.A.: Writing—review and editing (equal). O.K.D.: writing—review and editing (equal); conceptualization (supporting).
Declaration
Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture (USDA). USDA is an equal opportunity provider and employer.
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.