
Abstract
Vibrio parahaemolyticus is an important foodborne pathogen with diverse serotypes. In May 2021, we investigated a gastroenteritis outbreak that occurred in China, caused by V. parahaemolyticus O10:K4 infection. Based on the epidemiological curve, this outbreak was identified as a homologous exposure event. A case-control study demonstrated that emperor crab with mashed garlic (odds ratio [OR] = 4.60, p = 0.030; 95% confidence interval [95% CI]: 1.11–19.14), goose liver geoduck (OR = 4.50, p = 0.029; 95% CI: 1.12–18.13), shrimp (OR = 4.89, p = 0.021; 95% CI: 1.22–19.65), and sea cucumber (OR = 7.36, p = 0.005; 95% CI: 1.68–32.26) were the potential sources of the food poisoning. V. parahaemolyticus isolates from 18 laboratory-confirmed cases were all serotyped O10:K4, and determined to be sequence type ST3 via multilocus sequence typing. Pulsed field gel electrophoresis and whole-genome sequencing analysis revealed the identical pattern and 0–2 single nucleotide variation among these isolates. tdh was positive in all isolates, while trh and Orf8 were absent. Seven essential base positions in toxRS for pandemic clone identification were identical between the O10:K4 and O3:K6 pandemic clones. Phylogenetic analysis with 45 additional genomes of 13 different serotypes showed the closest genetic relationship between O10:K4 and O1: KUT. O10:K4 was thought to evolve from the O3:K6 pandemic clone. The new serovariant of O3:K6 poses a challenge for the prevention and control of V. parahaemolyticus disease outbreaks, or even epidemics, in the future.
Introduction
V
Although no definite correlation between serotypes and pathogenesis has been identified, certain serotypes have been considered main causes of outbreaks or even pandemics, for example, O3:K6 (Okuda et al., 1997; Chowdhury et al., 2000a), O1:K9 (Newton et al., 2012), O1:K25 (Chowdhury et al., 2000b; Matsumoto et al., 2000), O4:K64, O4:K68 (Chowdhury et al., 2000a), and O6:K18 (Newton et al., 2012). In 2004 and 2013, O4:K12 and O4: KUT caused large outbreaks in the United States (Martinez-Urtaza et al., 2013). Frequent genetic recombination has been observed in V. parahaemolyticus (Cui et al., 2015; Lu et al., 2016). If the recombination occurs around the O- and K-antigen-encoding gene cluster, the serotype might change (Chen et al., 2011). In this study, we investigated an outbreak of gastroenteritis caused by the emerging serotype O10:K4 of V. parahaemolyticus in China.
Materials and Methods
The outbreak
On May, 29, 2021, the emergency departments of three hospitals located in Huzhou City, Zhejiang Province, China, treated several patients with acute gastroenteritis. These patients reported having attended the same wedding banquet. This suspected outbreak of foodborne disease was subsequently reported to the market supervision department and health administrative authority. An investigation was conducted.
Case definition
A suspected case was defined as a person with abdominal pain, diarrhea, or vomiting after eating dishes at the wedding banquet. Diarrhea meant three or more episodes within 24 h. If two or more of the above symptoms occurred, a probable case could be determined, and could be defined as a laboratory-confirmed case if a stool sample from the patient was tested positive for V. parahaemolyticus.
Epidemiological investigation
The local public health physician interviewed the banquet attendees and physician-in-charge, and checked the outpatient and inpatient records to determine the cases. Probable cases were further investigated based on a standardized questionnaire. The questionnaire included basic demographic information (age and gender), clinical information (first symptoms, onset time, frequency of vomiting, abdominal pain and diarrhea, stool character, and body temperature) as well as food exposure. Cases (probable and confirmed) with symptoms developed on May 29–30 were included in a case-control study. Banquet attendees who had no gastroenteritis symptoms during this outbreak were defined as controls. On May 30, a detailed sanitary investigation was carried out on site, and kitchen environment samples were obtained for laboratory testing. However, because all the remaining food had been removed after the wedding banquet, food samples could not be collected on site.
Sample collection and bacteria isolation
Stool samples collected from probable case patients were forwarded for Salmonella spp., Shigella spp., V. parahaemolyticus, and enterotoxogenic Escherichia coli testing. Kitchen environment samples were tested for the same pathogens, as well as coliforms and total bacteria count. All detections were carried out according to the “National foodborne disease surveillance manual for 2021” (China National Center for Food Safety Risk Assessment, 2021). For V. parahaemolyticus, 3% sodium chloride alkaline peptone water was used for enrichment. After incubation at 36°C for 18 h, the cultures were plated on Chromogenic Vibrio agar (Chromogar, Paris, France) for presumptive colony screening. Presumptive colonies were further identified using the VITEK 2 compact system (Bio Mérieux, Lyon, France). Confirmed V. parahaemolyticus isolates were serotyped by slide agglutination, using commercial antisera (Denka Seiken, Tokyo, Japan).
Pulsed field gel electrophoresis
Pulsed field gel electrophoresis (PFGE) was carried out according to the recommended protocol (Xiao et al., 2014). Not I was used for total DNA restriction digestion. Electrophoresis was performed with the CHEF Mapper PFGE system (Bio-Rad Laboratories, Hercules, CA). Salmonella enterica serovar Braenderup digested with XbaI (Takara) was used as the reference marker and BioNumerics v. 7.6 software (Applied Maths, Kortrijk, Belgium) was used for Gel image analysis.
Genome sequencing, multilocus sequence typing, virulence genes, and pandemic gene-marker determination
The total DNA of V. parahaemolyticus isolates was extracted using Bacterial DNA Kit (Omega). Purified DNA was used for library preparation with TruePrep™ DNA Library Prep Kit V2. The prepared library was sequenced on the Illumina HiSeq X Ten platform (Illumina, Inc.) and de novo assembly was performed with CLC Genomics Workbench v10.0 (Qiagen, Hilden, Germany). The genome sequences have been deposited at DDBJ/ENA/GenBank under the accession numbers: JALEBF000000000, JALEBE000000000, JALEBD000000000, JALEBC000000000, JALEBB000000000, JALEBA000000000, JALEAZ000000000, JALEAY000000000, JALEAX000000000, JALEAW000000000, JALEAV000000000, JALEAU000000000, JALEAT000000000, JALEAS000000000, JALEAR000000000, JALEAQ000000000, JALEAP000000000, and JALEAO000000000.
For multilocus sequence typing (MLST), we submitted the genome sequence of each V. parahaemolyticus isolate to the PubMLST V. parahaemolyticus database (
The presence profiles of virulence genes tdh, trh, pandemic gene marker Orf8, and the sequence of toxRS was screened on V. parahaemolyticus genomes using BLAST 2.2.28+. We also performed polymerase chain reaction (PCR) for tdh, trh, and Orf8 detection (Laohaprertthisan et al., 2003; West et al., 2013) to confirm the whole-genome sequencing (WGS) findings. The discrepancies between the seven base positions in toxRS are essential for distinguishing O3:K6 old clones and new (pandemic) clones.
Therefore, to determine these discrepancies between O10:K4 and O3:K6 strains, we selected five representative O3:K6 new clone strains (VP81, JKY-VP6, BE-98-2062, VP108, and FIHES98V14-1) and four representative O3:K6 old clone strains (U-5474, AQ4901, AQ3810, and DOH272) (Matsumoto et al., 2000). The toxRS nucleotide sequences of the above strains were obtained from the GenBank database, and had the following accession numbers: AB029911 (VP81), AB029913 (JKY-VP6), AB029903 (BE-98-2062), AB029912 (VP108), AB029904 (FIHES98V14-1), AB029915 (U-5474), AB029909 (AQ4901), AB029907 (AQ3810), and AB029908 (DOH272). Multialignment was performed using ClustalX 1.83.
We selected the genome sequence of V. parahaemolyticus isolate 21–215 as a reference. We then mapped genome raw data for 17 other V. parahaemolyticus isolates in this study, and 45 additional V. parahaemolyticus strains belonging to different serotypes (including O1:K25, O1:K56, O1:K1, O1: KUT, O3:K59, O3: KUT, O4:K8, O4:K12, O4:K11, O4:K63, O4:K68, O3:K6, and O5:KUT) retrieved from the NCBI GenBank SRA database to the reference and aligned them using the Snippy pipeline (
Antibiotic susceptibility tests
Antibiotic susceptibility of all V. parahaemolyticus isolates was assayed using the broth microdilution minimum inhibitory concentrations method according to the Clinical and Laboratory Standards Institute guidelines (Institute CLSI, 2015). We used the breakpoints for Cefoxitin, Azithromycin, Chloramphenicol, Tetracycline, Amoxicillin-clavulanate, Ciprofloxacin, Gentamicin, Trimethoprim-Sulfamethoxazole, and Ampicillin in CLSI documents M45-A3 (Institute CLSI, 2015). E. coli ATCC29522 was used as a quality-control strain.
Statistical analysis
Descriptive analysis was performed with SPSS 23.0 (SPSS, Inc.). The chi-square test or Fisher's test was used to compare qualitative variables. The case-control odds ratio (OR) and 95% confidence intervals (95% CIs) were calculated to measure the association between exposure and outcome. Statistical significance was defined as p < 0.05.
Results
Epidemiological investigation
Fifty-eight suspected cases, 28 probable cases, and 15 laboratory-confirmed cases were determined in this outbreak among 500 banquet attendees, respectively. The attack rate was 5.60% (28/500). The first case, a female patient, aged 62, began to have abdominal pain and diarrhea (seven times, mainly watery stool) at 18:00 on May 29, accompanied by nausea and vomiting. In all probable cases, the main clinical symptoms were abdominal pain and diarrhea, accompanied by nausea and vomiting, and diarrhea was mostly watery stool. Most cases had diarrhea 5–10 times a day. Ten cases had fever, and the details are shown in Table 1. The patients received emergency treatment in local hospitals. After giving symptomatic support treatment, including rehydration and antidiarrheal medication, they were all cured without any severe cases or deaths.
Clinical Symptoms of 28 Probable Cases
The initial case occurred at 18:00 on May 29, and the final case occurred at 7:00 on May 30. The incidence peak presented at 2:00 on May 30, and the time between the first case and the last case was about 13 h. According to the epidemiological curve, this outbreak was a homologous exposure event. The suspicious meal was the lunch on May 29 (at 12 o'clock). The incubation period ranged from 6 to 19 h, and the average incubation period was about 13 h. The specific distribution of onset time is shown in Figure 1.
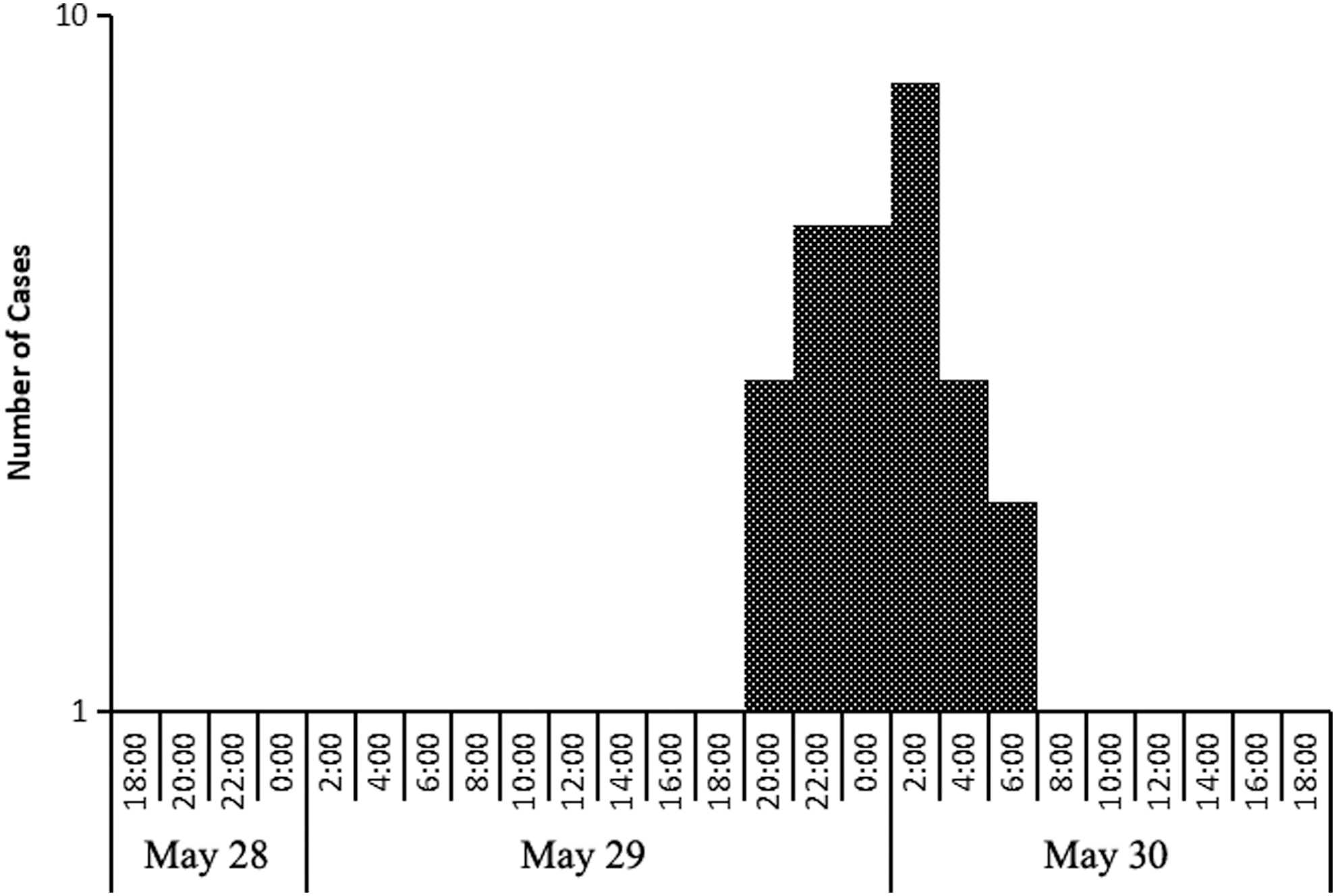
The onset time distribution of 28 probable cases.
Among the 28 possible cases, the youngest patient was 7 years old and the oldest was 78 years old. There were 13 males and 15 females.
Case-control study
To further identify suspicious food, we carried out a case-control study with 28 possible cases and 14 controls enrolled. Considering that the food consumption of cases and controls at the wedding banquet could not be recalled clearly, only seafood consumption was investigated during the case-control investigation. Univariate analysis indicated that consumption of emperor crab with mashed garlic (OR = 4.60, p = 0.030; 95% CI: 1.11–19.14), goose liver geoduck (OR = 4.50, p = 0.029; 95% CI: 1.12–18.13), shrimp (OR = 4.89, p = 0.021; 95% CI: 1.22–19.65), and sea cucumber (OR = 7.36, p = 0.005; 95% CI: 1.68–32.26) were significantly different between the case and control groups. These four dishes were therefore potential sources of food poisoning. Food consumption of the case and control groups on May 29 is presented in detail in Table 2.
Seafood Consumption of Case Group and Control Group on May 29
95% CI, 95% confidence interval; OR, odds ratio.
Laboratory results
Stool samples collected from 20 possible cases were all negative for Salmonella spp., Shigella spp., and enterotoxogenic E. coli. Eighteen of the samples were identified as V. parahaemolyticus-positive, with a detection rate of 90%. One V. parahaemolyticus strain was isolated from each positive sample. A serotyping test demonstrated that all 18 V. parahaemolyticus isolates belonged to O10:K4. In addition, four kitchen environmental samples were negative for Salmonella spp., Shigella spp., V. parahaemolyticus, and enterotoxogenic E. coli. Three of these were coliform positive.
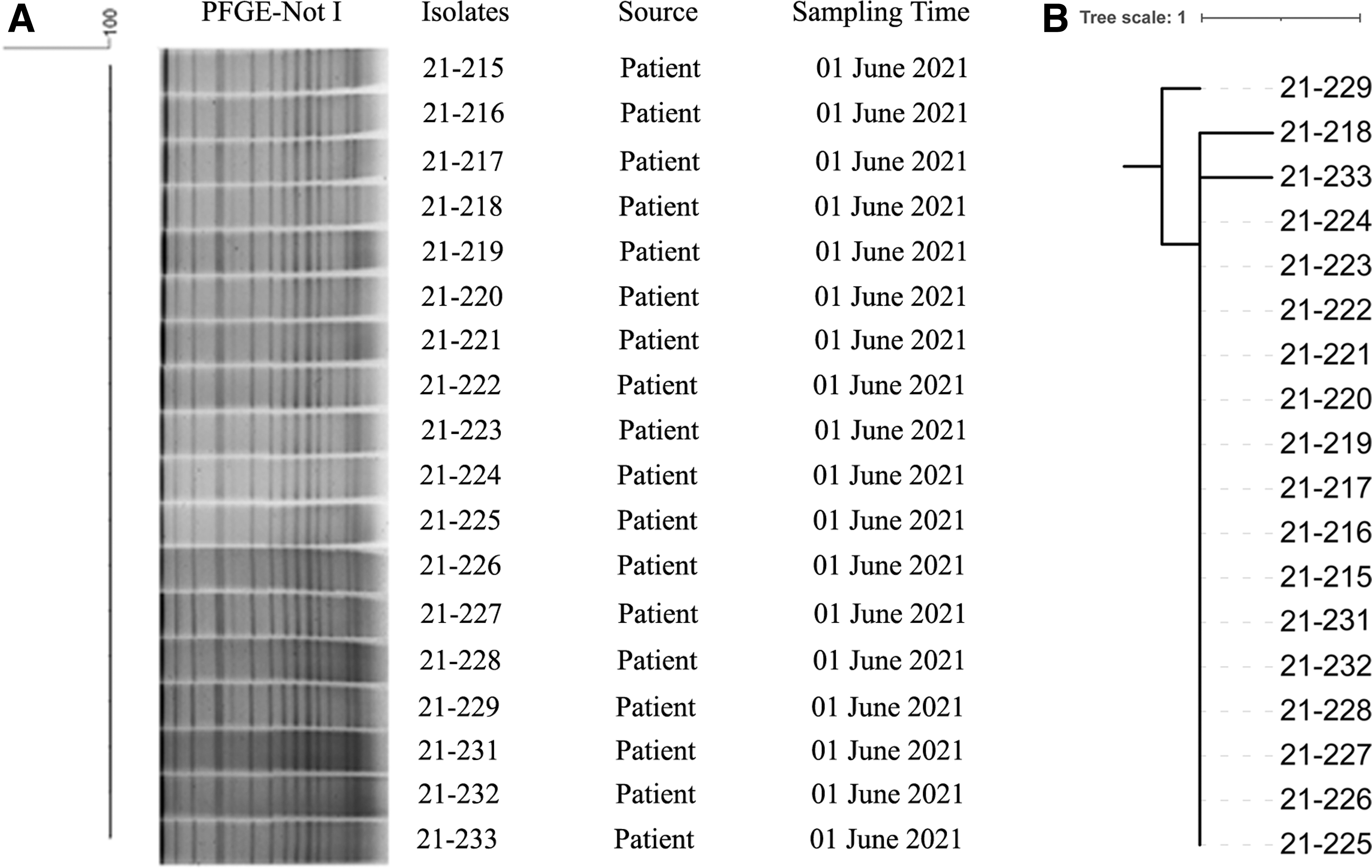
V. parahaemolyticus isolates were further typed with PFGE (Fig. 2A). All isolates presented 100% similarity. WGS analysis demonstrated that all 18 isolates differed in 0–2 SNPs (Fig. 2B). To understand the genetic relationship of the emerging serotype O10:K4 with other serotypes, we performed phylogenetic analysis with additional 45 genomes of 13 serotypes retrieved from the NCBI GenBank. O10:K4 isolates showed the closest genetic relationship with V. parahaemolyticus strain AP-11243 and strain AN-16000 (1747 to 1833 SNPs). These two strains were isolated in Bangladesh in 1998 and 2000, respectively, and clustered into one subclade 2.2.2.2.2.2.2 (Fig. 3). These two isolates both belonged to serotype O1:KUT, which has been identified as one serovariant of the O3:K6 pandemic clone (Matsumoto et al., 2000; Bhuiyan et al., 2002). We also found that strains of O10:K4, O3:K6, and O3:K6 serovariants, including O4:K68, O1:K25, and O1: KUT, were distributed in the same subclade 2.2.2.2.
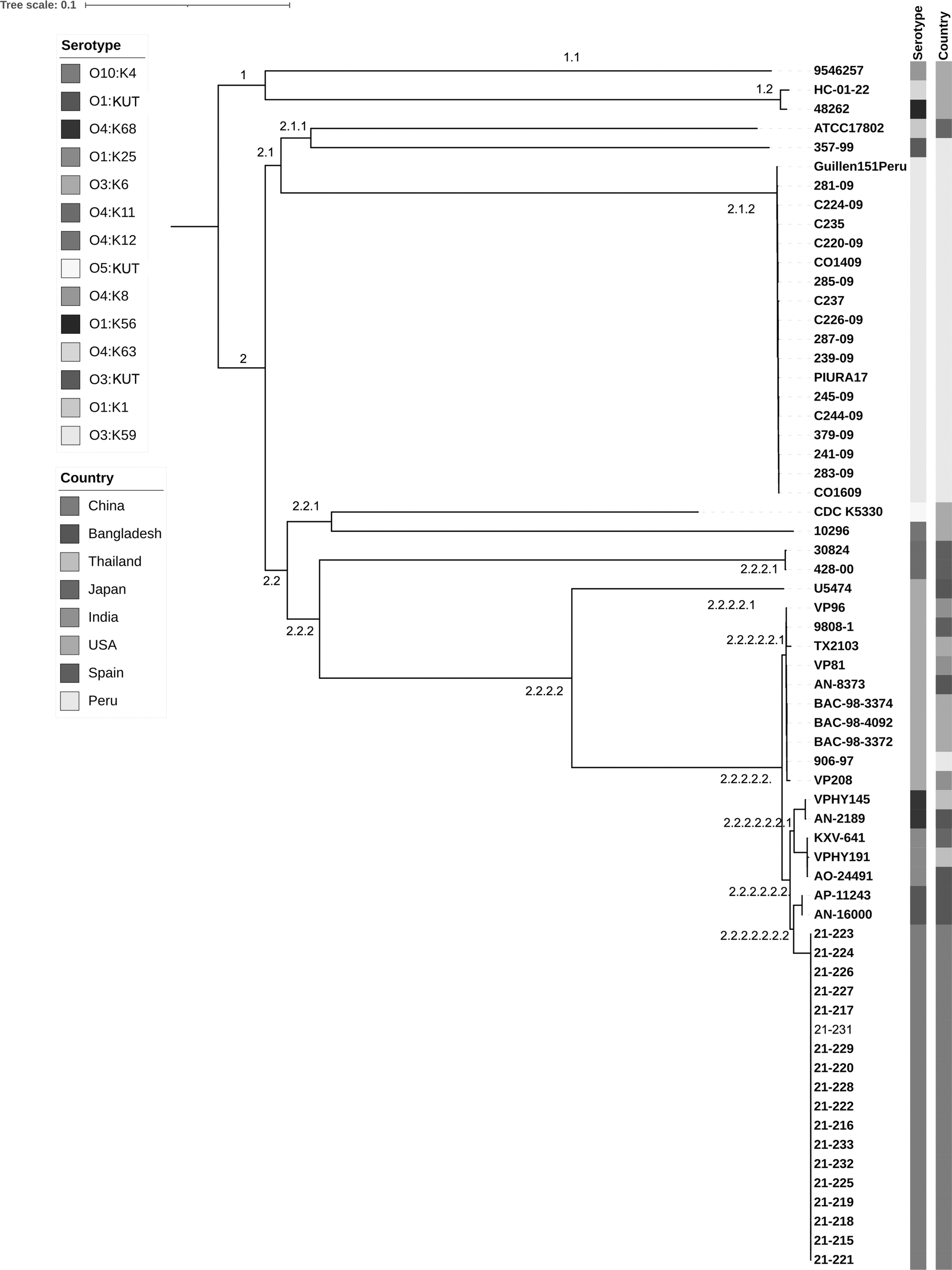
Genome phylogeny of Vibrio parahaemolyticus strains with various serotypes, including O1:K25, O1:K56, O1:K1, O1:KUT, O3:K59, O3:KUT, O4:K8, O4:K12, O4:K11, O4:K63, O4:K68, O3:K6 O5::KUT, and O10:K4. A total of 45 genomes representing a data set of major serotypes detected from clinical cases were retrieved from GenBank, and included in genome SNPs analysis along with 18 V. parahaemolyticus strains isolated in this study. SNP, single-nucleotide polymorphism.
Through alignment between genome sequences and reference MLST alleles, we were able to determine that all V. parahaemolyticus isolates were of the same ST, ST3. tdh was present in all isolates, whereas both trh and Orf8 were absent. The presence profiles of tdh, trh, and Orf8 were also confirmed using PCR. Furthermore, the V. parahaemolyticus O10:K4 isolates reported here possessed the same seven base positions located in toxRS to O3:K6 new clones (pandemic clones) (Matsumoto et al., 2000). These bases were A576, A900, T1002, T1196, T1214, A1244, and T1463 (Fig. 4).
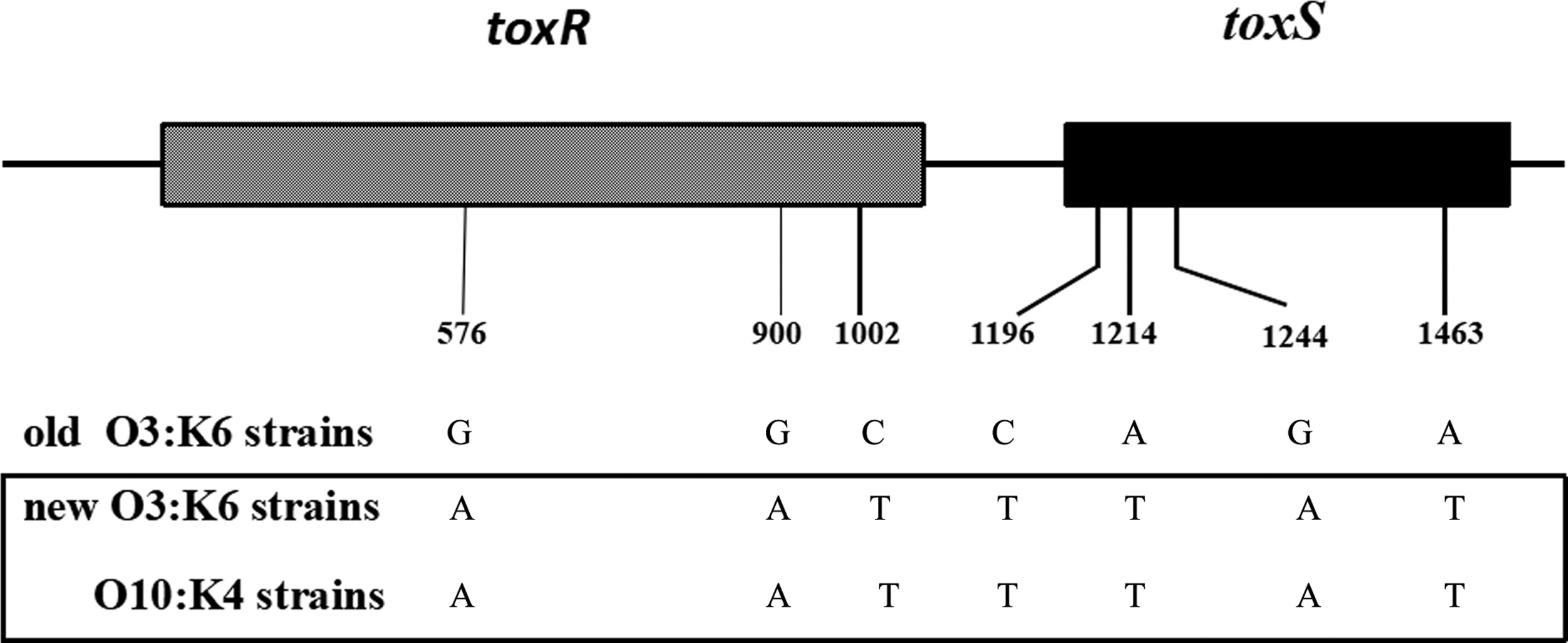
The essential seven bases difference in the toxRS sequence among old O3:K6 strains, new O3:K6 strains, and O10:K4 strains.
The susceptibility of V. parahaemolyticus isolates to nine antibiotics is shown in Table 3. All 18 strains were susceptible to Azithromycin, Tetracycline, Gentamicin, Chloramphenicol, Ciprofloxacin, and Trimethoprim-sulfamethoxazole. 11.1%, 38.9%, and 88.9% of isolates were susceptible to Ampicillin, Cefoxitin, and Amoxicillin-clavulanate, respectively. V. parahaemolyticus isolates presented resistance to three types of antibiotics: Ampicillin, Cefoxitin, and Amoxicillin-clavulanate. Of these, the predominant antibiotic resistance type was resistance to Ampicillin. Of V. parahaemolyticus isolates, 88.9% were resistant to this antibiotic. Three isolates were resistant to Cefoxitin and two were resistant to Amoxicillin-clavulanate.
Antibiotic Susceptibility of V. parahaemolyticus Isolates
MIC, minimum inhibitory concentration.
Discussion
Before 1996, although the regional dominance of a specific serotype of V. parahaemolyticus had occurred occasionally, for example, the dominance of serotype O1:K56 among sporadic diarrheal cases in Calcutta (Chatterjee and Sen, 1974) and of O4:K12 along the western coasts of Mexico and the United States (Abbott et al., 1989), no specific serotype was associated with disease outbreaks or even epidemic. During that year, the first O3:K6 caused a cluster of cases in Calcutta. Unlike the previously reported clustering of V. parahaemolyticus serotypes, O3:K6 rapidly increased hospitalizations and became the predominant serotype in some areas (Yamazaki et al., 2003; Nair et al., 2007). This outbreak rapidly spread globally, leading to infection of thousands of people with V. parahaemolyticus (Nair and Hormazabal 2005; Nair et al., 2007).
During O3:K6 dissemination, molecular analyses revealed that the newly emerged clone had diversified into more than 20 serovariants. O3:K6 and its serovariants were termed the “pandemic clones.” The pandemic clones are genetically similar with regard to ribotyping, arbitrarily primed (AP)-PCR, PFGE, group-specific (GS)-PCR for toxRS, and ORF8 PCR for the f237 filamentous phage, regardless of their diverse serotypes (Chowdhury et al., 2000b; Nasu et al., 2000). The toxR and toxS genes in the toxRS operon encode two conserved transmembrane proteins involved in the regulation of virulence genes in the genus Vibrio (DiRita, 1992; Lin et al., 1993).
O3:K6 strains (old clone) isolated before February 1996, when a group of O3:K6 strains (pandemic clone, new clone) possessing the tdh gene, but not the trh gene, appeared suddenly in Calcutta and caused global V. parahaemolyticus epidemic, have different toxRS sequence from that of the new clone at 7 base positions within a 1346-bp region. GS-PCR targeting two of the base positions unique to the O3:K6 new clone was developed to specifically detect the O3:K6 pandemic clone and its serovariants (Matsumoto et al., 2000). Phage f237 of V. parahaemolyticus is similar to the CTX filamentous phage of Vibrio cholerae O1 (Waldor and Mekalanos, 1996). ORF8 of phage f237 is thought to play a significant role in enhancing the virulence of O3:K6 pandemic clones.
Nasu et al. (2000) presumed that ORF8 encodes an adherence protein that could increase the adhesion capability to host intestinal cells or the surfaces of marine plankton. The presence or absence of ORF8 in V. parahaemolyticus strains has become a rapid indicator for pandemic clone identification. In addition, MLST data have confirmed that O3:K6 and several serovariants occur in a single genetic lineage (Chowdhury et al., 2004a). Meanwhile, O3:K6 and its serovariants have identical virulence gene genotype: tdh positive and trh negative (Okuda et al., 1997).
V. parahaemolyticus isolates from the gastroenteritis outbreak reported in this study were all serotyped O10:K4. An identical PFGE pattern and few SNPs (≤2) revealed that these isolates were highly homologous and possibly from the same origin. Molecular analysis further demonstrated that this new serotype had common characteristics with the O3:K6 pandemic clone. tdh existed in all isolates, whereas trh was absent. The seven base positions of toxRS, essential for distinguishing the O3:K6 old clone and pandemic clone, were identical in O10:K4 strains and in the O3:K6 pandemic clone. Meanwhile, ORF8 was not detected in O10:K4 strains. Previously published reports have revealed inconsistencies between the results for toxRS and ORF8 PCRs. Some other serotypes that were not like the O3:K6 pandemic clone also had ORF8 (Iida et al., 2001; Osawa et al., 2002; Chowdhury et al., 2004b). Both toxRS and ORF8 were unreliable for definite identification of the pandemic group.
To resolve the confusion caused by the inconsistent detection results of pandemic clone gene markers for the O10:K4 isolates, we performed phylogenetic analysis based on genome SNPs. O10:K4 isolates presented the closest genetic relationship with O1:KUT isolates, identified as a O3:K6 serovariant from Bangladesh in 1998 and 2000. Furthermore, O10:K4 isolates clustered with O3:K6 and its serovariants into one subclade. Based on the combination of gene marker detection results and genome SNP phylogenetic analysis, we speculate that O10:K4 evolved from the O3:K6 pandemic clone, and that it is one of the serovariants.
Besides gene markers, PFGE, MLST, AP-PCR, and ribotyping are usually utilized as general methods for pandemic group identification (Chowdhury et al., 2000a, b; Wong et al., 2000). MLST and ribotyping, however, are targeted to partial genetic information, and thus may give indefinite results. PFGE and AP-PCR can provide whole-genome profiles, but band patterns make it difficult to compare different bacteria strains in different laboratories, and comparison results may be subjective. The availability of genome sequencing provides a more efficient method for genetic analysis and makes pandemic group identification more reliable and objective.
Due to the lack of food samples and the negative V. parahaemolyticus results from kitchen environmental samples, the source of food poisoning in this outbreak remains unclear. To explore the potential food source of the new V. parahaemolyticus serotype O10:K4, we screened food-microorganism surveillance data from Zhejiang Province from 2011 to September 2021. No O10:K4 isolates were detected in any of the food samples. Emergence of a new serovariant of the pandemic clone may be a response to host immunological pressure, according to a previous report (Chowdhury et al., 2004a). O10:K4, the new serovariant of O3:K6, poses a challenge for the prevention and control of V. parahaemolyticus disease in the future.
Ethics
This study was approved by the Ethics Committee of the Zhejiang Provincial Center for Disease Control and Prevention. All the data of the individuals were kept confidential as requested.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Key Research and Development Program of China (2017YFC1601503) and The Natural Science Foundation of Zhejiang Province (LY20H190001).