
Abstract
The microbes on fresh processing tomatoes correlate closely with diseases, preservation, and quality control. Investigation of the microbial communities on processing tomatoes from different production regions may help define microbial specificity, inform disease prevention methods, and improve quality. In this study, surface microbes on processing tomatoes from 10 samples in two primary production areas of southern and northern Xinjiang were investigated by sequencing fungal internal transcribed spacer and bacterial 16S rRNA hypervariable sequences. A total of 133 different fungal and bacterial taxonomies were obtained from processing tomatoes in the two regions, of which 63 genera were predominant. Bacterial and fungal communities differed significantly between southern and northern Xinjiang, and fungal diversity was higher in southern Xinjiang. Alternaria and Cladosporium on processing tomatoes in southern Xinjiang were associated with plant pathogenic risk. The plant pathogenic fungi of processing tomatoes in northern Xinjiang were more abundant in Alternaria and Fusarium. The abundance of Alternaria on processing tomatoes was higher in four regions of northern Xinjiang, indicating that there is a greater risk of plant pathogenicity in these areas. Processing tomatoes in northern and southern Xinjiang contained bacterial genera identified as gut microbes, such as Pantoea, Erwinia, Enterobacter, Enterococcus, and Serratia, indicating the potential risk of contamination of processing tomatoes with foodborne pathogens. This study highlighted the microbial specificity of processing tomatoes in two tomato production regions, providing a basis for further investigation and screening for foodborne pathogenic microorganisms.
Introduction
Tomato (Solanum lycopersicum L.) is one of the most widely cultivated plants worldwide that is well regarded for its high nutritional value (John, 2008). Processing tomato is a cultivated type of common tomatoes that received its name from its thick skins, which is resistant to transport damage and suitable for processing (Qin et al., 2022). Tomato processing is one of Xinjiang's red industries, which was proposed as a collective term for Xinjiang's red agricultural products (e.g., safflower carthamus, chilis, tomatoes, goji berries, jujubes etc.) and their processing industries. A total export volume of 1.028 million tons of product is exported to multiple regions across Europe, America, Africa, and Central Asia (Yao et al., 2021). However, processing tomatoes are susceptible to several foodborne diseases, not only resulting in loss during production and storage, but also being harmful to human beings (Zhou et al., 2019).
Foodborne diseases mainly refer to infectious and toxic diseases caused by ingestion of food contaminated by biological pathogens and toxic and harmful chemicals (Sun, 2021). Foodborne pathogenic microorganisms are the main biological hazards that contaminate food and edible-agricultural products and seriously threaten the health of humans and animals. At the same time, they also cause huge economic losses to food and edible agricultural products industry, which is the key factor of restricting their development.
Foodborne diseases caused by bacterial contamination of food account for the vast majority. It is estimated that foodborne pathogenic bacteria are responsible for ∼45% of food safety events worldwide each year (Ghadeer et al., 2017). There are some more common foodborne pathogenic bacteria that tend to cause some serious events, including the following pathogens, Staphylococcus aureus, Salmonella, Listeria monocytogenes, Shigella, E. coli O157:H7, and Vibrio parahaemolyticus (Wang et al., 2022).
Fresh fruits and vegetables can accommodate large and diverse populations of fungi and bacteria. However, most of the work on the production of isolated bacteria has focused on relatively few pathogenic bacteria (Zoellner et al., 2016). To date, little is known about the overall diversity and composition of microbial communities in fruits and vegetables, especially on processing tomatoes. Thus, there is a critical need for a robust systematic study of the microbial communities in processing tomato.
In recent years, microbiome sequencing technology has been used in many fields, including the sequencing and analysis of plant (Hartmann et al., 2019), plant root (Cui et al., 2022), human intestinal (Nuli et al., 2019), and soil microorganisms (Yang et al., 2020). Until recently, metagenomic analysis of microbial ribosomal DNA (rDNA) hypervariable region sequences has allowed for breakthroughs in the study of microbial communities in various biological samples (Taîbi et al., 2021; Taylor et al., 2014).
However, microbial communities associated with processing tomatoes have not been systematically studied using microbiome research technology. This study assessed the microbial communities on processing tomatoes from primary production areas in different regions of Xinjiang. The fungal internal transcribed spacer (ITS) and the bacterial 16S rRNA hypervariable region were sequenced to study the composition of microbial community structure after harvest. The findings provide a theoretical basis to clarify the mechanism of tomato deterioration from the perspective of endophyte and epiphytic microorganisms.
This paper intends to provide the latest references or innovative ideas for the screening and traceability of foodborne pathogenic microorganisms in processing tomatoes, which is also of great significance to the healthy development of the processing tomato industry in Xinjiang.
Materials and Methods
Sample collection and microbial DNA extraction
In 2021, processing tomatoes were picked from 10 main production areas (Fig. 1). During sampling, 5 kg of tomato fruits was collected using the five-point sampling method. Within 2 days, the tomatoes were protected with breathable mesh bags and transported to the laboratory. From each area, 1 kg of tomato samples was randomly selected from 5 kg of tomatoes, broken into tomato pulp using a juicer, and mixed well, and 0.25–0.5 g of tomato homogenate was weighed for DNA extraction.

The location of processing tomatoes sampled from northern and southern Xinjiang, China. (NJ-FQ1–2 represent the sample numbers of Hejing County in southern Xinjiang located in the middle and upper left of the map; NJ-FQ3–5 represent the sample numbers of Bohu County, Yanqi County, and Heshuo County in southern Xinjiang, respectively; and BJ-FQ1–5 represent the sample numbers of Shihezi City, Manas County, Changji City, Jimsal County, and Usu City in northern Xinjiang, respectively.)
Total genomic DNA samples were extracted using the DNeasy® PowerSoil® Kit (QIAGEN, Inc., Netherlands), following the manufacturer's instructions, and stored at −20°C before further analysis. The quantity and quality of extracted DNAs were measured using a NanoDrop NC2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA) and agarose gel electrophoresis, respectively.
PCR amplification and Illumina sequencing
The hypervariable region V5–V7 of the bacterial 16S rRNA gene was amplified using the primer pairs, 779F (5′-AACMGGATTAGATACCCKG-3′) and 1193R (5′-ACGTCATCCCCACCTTCC-3′) (Horton et al., 2014), with an ABI GeneAmp® 9700 PCR thermocycler (ABI, CA). The fungal primers were ITS1F: (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS2: (5′-GCTGCGTTCTTCATCGATGC-3′) (Orgiazzi et al., 2012).
The PCR mixture included 5 μL 5 × reaction buffer, 2 μL 2.5 mM dNTPs, 1 μL of 10 μM forward primer, 1 μL of 10 μM reverse primer, 0.25 μL Q5 DNA polymerase, 2 μL DNA template, and ddH2O to a final volume of 25 μL. PCR amplification cycling conditions were as follows: initial denaturation at 98°C for 2 min, followed by 27 cycles of denaturing at 98°C for 15 s, annealing at 55°C for 30 s, extension at 72°C for 30 s, single extension at 72°C for 5 min, and storage at 4°C. All samples were amplified in triplicate. The PCR product was recovered from a 1.2% agarose gel, purified using the AxyPrep DNA gel extraction kit (Axygen Biosciences, Union City, CA) according to the manufacturer's instructions, and quantified using a Quantus™ fluorometer (Promega).
Illumina MiSeq sequencing
Purified amplicons were pooled in equimolar amounts and paired-end sequenced using an Illumina MiSeq PE300 platform (Illumina, San Diego) by Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China) according to the standard protocols. The raw sequencing reads were deposited into the NCBI Sequence Read Archive database (Accession No.: PRJNA885764).
Data processing
Raw FASTQ files were de-multiplexed using an in-house perl script, quality-filtered using fastp version 0.19.6 (Chen et al., 2018), and merged using FLASH version 1.2.7 (Magoč and Salzberg, 2011). The optimized sequences were clustered into operational taxonomic units (OTUs) using UPARSE 7.1 (Edgar and Robert, 2013) with 97% sequence similarity. The most abundant sequence for each OTU was selected as a representative sequence.
To minimize the effects of sequencing depth on alpha and beta diversity measure, the number of 16S rRNA and ITS gene sequences from each sample was rarefied to 41,883 and 112,302, which still yielded an average Good's Coverage of 99.09%, respectively. The taxonomy of each OTU representative sequence was analyzed using RDP Classifier version 2.2 (Wang et al., 2007) against the 16S rRNA gene database (e.g., Silva v138) using a confidence threshold of 0.97.
The database of fungal ITS sequences is the UNITE database (Release 8.0,
Statistical analysis
Bioinformatic analysis of the processing tomato was performed using the Personal Cloud platform. Based on the OTU information, rarefaction curves and alpha diversity indices, including observed OTUs, Chao1 richness, Shannon index, and Good's Coverage, were calculated using QIIME software (Chao, 1984; Shannon, 1948; v1.8.0). The Kruskal–Wallis test was used to analyze differences in Alpha diversity between the groups.
Using QIIME and R software, OTU distributions at the phylum, class, order, family, and genus levels were examined. To assess variations in microbial composition between samples, beta diversity analyses were performed. To separate samples, partial least squares discrimination analysis (PLS-DA) and principal component analysis (PCA) were used.
Using the online Galaxy workflow framework, linear discriminant analysis (LDA score >2, p < 0.05; Segata et al., 2011) and linear discriminant effect size (LEfSe) analysis were performed to identify taxonomies that differed between groups. Index and microbial composition differences between samples were determined using a two-tailed Student's t-test (p < 0.05).
Results
Overall microbial community on processing tomatoes
In the fungal bacterial analysis, processing tomatoes had 112,302–139,952 high-quality ITS sequences, with an average of 126,885 sequences. The ITS reads were then aggregated into 23–125 OTUs, averaging 72 OTUs (Table 1). The predominant fungal taxonomies (relative abundance ≥0.05%) contained 3 phyla, 10 classes, 15 orders, 19 families, and 20 genera (Table 2).
High-Quality Fungal and Bacterial 16S rDNA V5–V7 Sequences, Clustered Operational Taxonomic Unit Numbers, and Genera of Processing Tomatoes from 10 Regions
ITS, internal transcribed spacer; OTU, operational taxonomic unit.
Fungi and Bacteria Taxonomies in Predominant Classification Levels (Relative Abundance ≥0.05%) and Their Percentages on Processing Tomatoes from 10 Regions
In the fungal taxa, Ascomycota phylum was dominant in the fungal community, followed by Basidiomycota phylum. Vishniacozyma had a high relative abundance in NJ-FQ4 and BJ-FQ5 of 58.69% and 41.58%, respectively, and Fusarium had a high relative abundance in BJ-FQ3 of 37.16%. Cladosporium had the highest abundance in NJ-FQ4 at 7.54%. Alternaria was the most common genus found on processing tomatoes in 10 regions, with a relative abundance of 97.02% in NJ-FQ3 (Fig. 2).
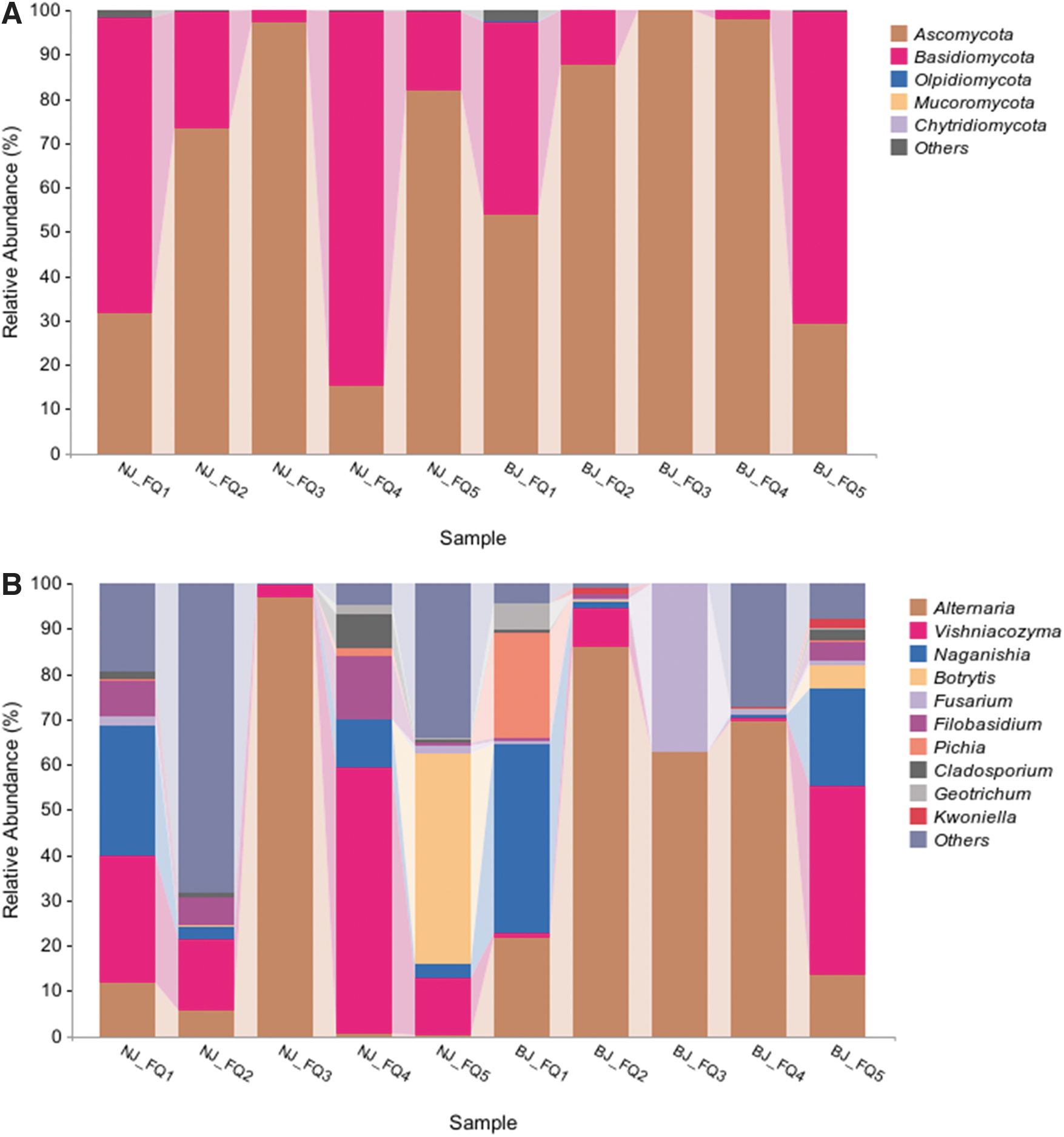
Fungal community structure of processing tomato at the phylum
In the bacterial analysis, processing tomatoes were observed to have 41,883–183,617 high-quality 16S rRNA reads, with an average of 69,682 reads. The reads clustered between 325 and 1511 OTUs, averaging 730 OTUs (Table 1). The predominant bacterial taxonomies (relative abundance ≥0.05%) included 7 phyla, 12 classes, 27 orders, 52 families, and 73 genera (Table 2). The relative abundance of Proteobacteria in NJ-FQ3, BJ-FQ3, and BJ-FQ4 was 66.46%, 78.47%, and 70.40%, respectively.
The Proteobacteria phylum mainly contained the genera of Pantoea, Pectobacterium, Erwinia, Enterobacter, Enterococcus, Xanthomonas, Pseudomonas, and Serratia. The highest relative abundance of Pantoea was present in BJ-FQ4 at 48.07%. The highest relative abundance of Pectobacterium was present in BJ-FQ3 at 50.11%. The highest relative abundance of Erwinia was found in NJ-FQ3 at 24.37%.
The highest relative abundance of Enterobacter was found in NJ-FQ3 at 17.33%. The highest relative abundance of Enterococcus was found in BJ-FQ4 at 11.54%. The common genera found in processing tomatoes in the 10 study regions were Pantoea, Enterobacter, Kocuria, and Pseudomonas (Fig. 3). There were also many unknown bacterial communities in processing tomato samples.
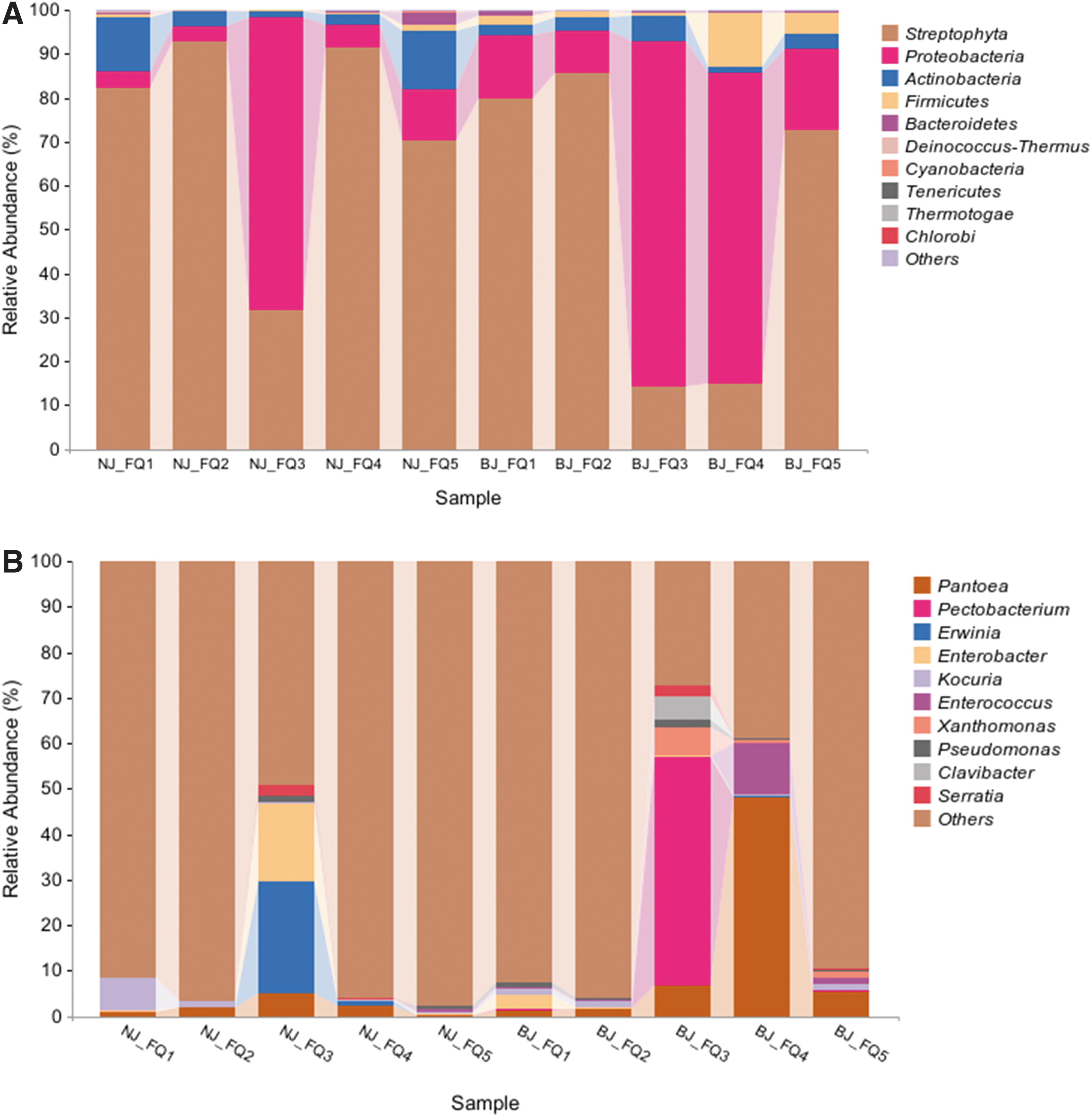
Bacterial community structure of processing tomato at the phylum
The alpha and beta diversity of microbial communities
Alpha diversity analysis revealed the diversity of microbial richness and evenness, including the abundance grade curve, species accumulation curve, and alpha diversity index. The OTU abundance rank curve began with a steep slope, followed by a long, flat tail (Fig. 4A) that grew larger and larger along the horizontal axis. This was indicative of an increase in fungal species richness in processed tomato samples from these 10 regions.
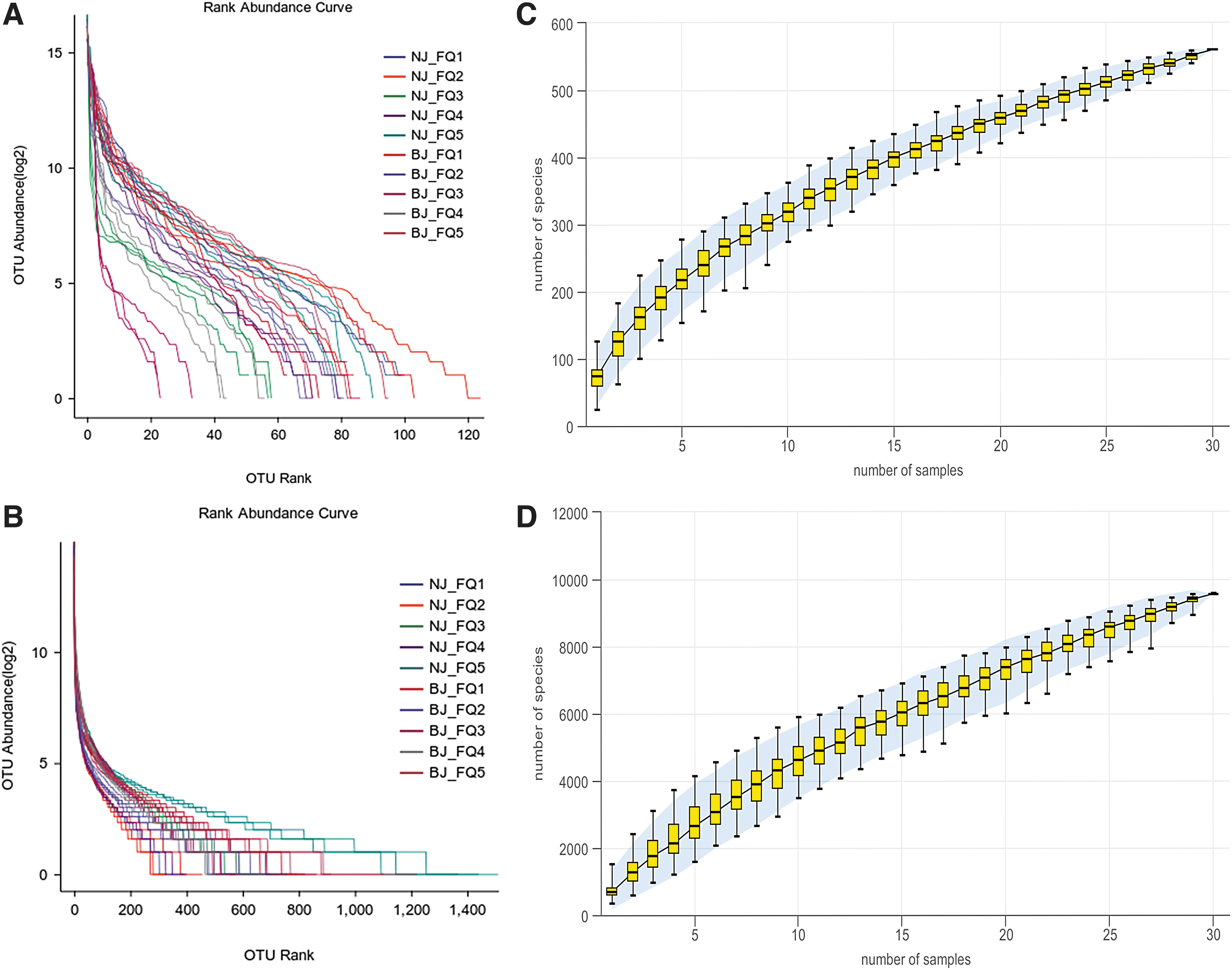
Abundance grade curves of fungi
The OTU abundance rank curve and the species accumulation curve (Fig. 4) of the primary samples were similar in shape, indicating that they had similar microbial richness and evenness. Both curves also showed adequate sequencing depth. The Simpson and Shannon diversity indexes of processing tomatoes were higher in southern than in northern Xinjiang (Fig. 5), indicating that processing tomatoes in southern Xinjiang had richer and more diverse fungal communities.

Box plots of alpha diversity index of fungi
Beta diversity analysis showed that the microorganisms associated with processing tomatoes contained significantly different microorganisms. PCA reduced the data dimension to visualize microbial differences. PCA of fungi showed that the contribution rates of principal component analysis 1 (PC1) and principal component analysis 2 (PC2) reached 57% and 18.3%, respectively, for a total of 75.3% (Fig. 6A). PLS-DA successfully distinguished between the processing tomato samples from different regions (Fig. 6C).
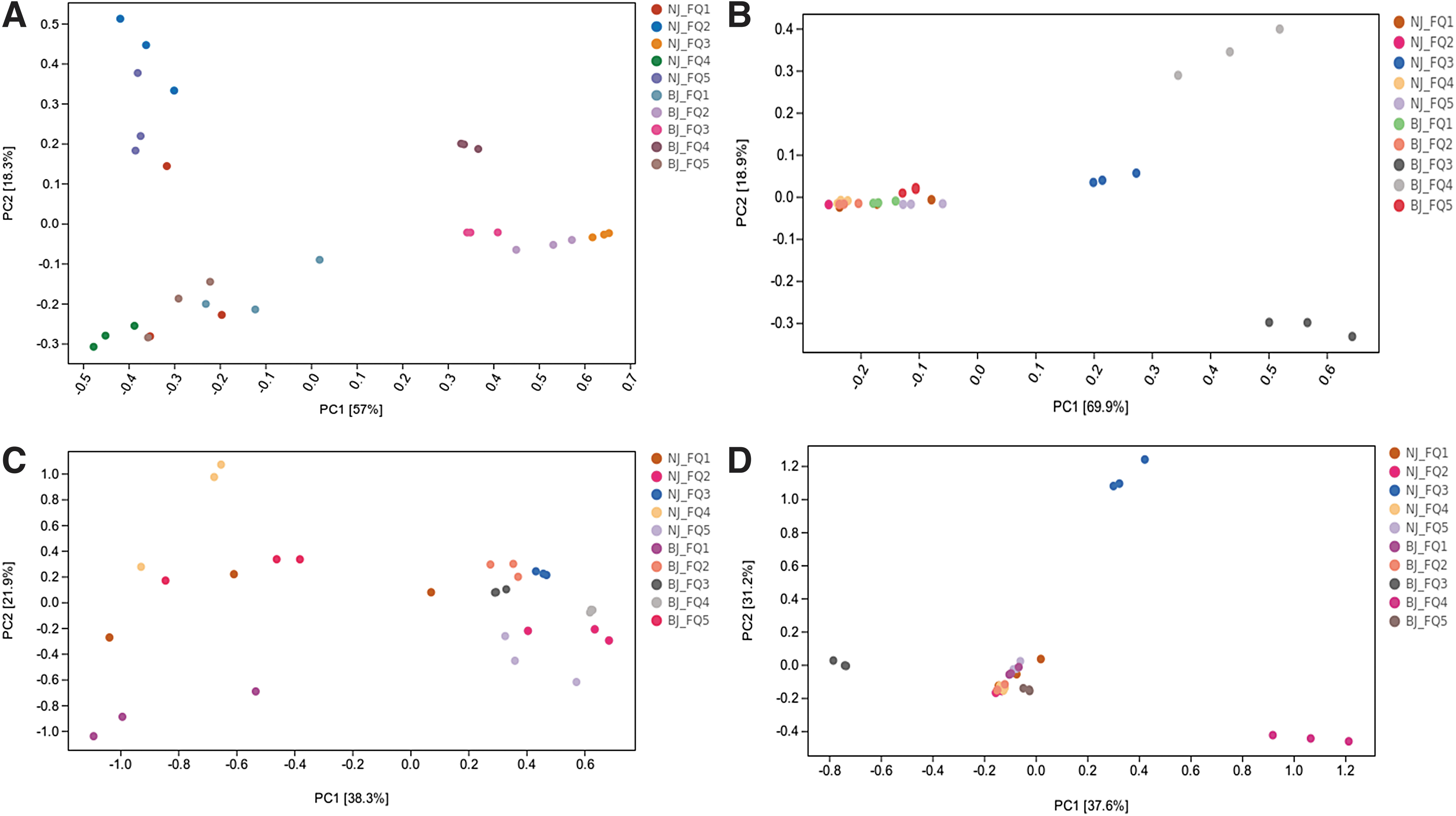
PCA of fungal
PCA of bacteria (Fig. 6B) showed that PC1 and PC2 explained 69.9% and 18.9% of the variability, respectively, for a total of 88.8% variation. It can be seen that the diversity of processing tomato fungal communities in the northern and southern Xinjiang regions differed greatly, and also varied among different samples from the same region; whereas the diversity of bacterial communities in the two samples from northern Xinjiang differed greatly from those from southern Xinjiang, and the different samples from southern Xinjiang had a high degree of similarity in bacterial species.
Bacterial community species similarity was high. In addition, while there was considerable overlap in the bacterial PLS-DA map, there was a clear separation between NJ-FQ3, BJ-FQ3, and BJ-FQ4 and the remaining seven samples (Fig. 6D). These findings showed that PCA and discriminant analysis could distinguish between different processing tomato production regions, indicating that there were significant differences in the fungal and bacterial communities on tomatoes found in different regions.
However, in the present study, PERMANOVA analysis was used to compare fungal and bacterial samples of processing tomatoes from different regions. It was found that there was no significant difference in species composition and relative abundance between the different taxa (p > 0.05).
Comparative analysis of the microbial communities
LEfSe analysis is a method that combines nonparametric Kruskal–Wallis and Wilcoxon rank sum tests with LEfSe. LEfSe analysis can also be used to simultaneously assess the differences between all classification levels. This method finds robust between-group microbial species differences that can be used as biomarkers.
LEfSe was used to identify different fungal and bacterial classifications between the two groups (LDA ≥2.0 and p ≤ 0.05). A total of 39 different fungal taxa were obtained from 10 groups of processing tomatoes, including 2 phyla, 6 classes, 10 orders, 10 families, and 11 genera (Fig. 7A). The longer the bar chart, the more significant the difference in the taxonomic units. Dothideomycetes, Pleosporales, Pleosporaceae, and Alternaria were shown to differ significantly in NJ-FQ3.

LEfSe analysis of different fungal
There were significant differences in 94 bacterial taxa among the 10 groups, including 2 phyla, 3 classes, 14 orders, 23 families, and 52 genera (Fig. 7B). Viridiplantae and Streptophyta differed significantly in NJ-FQ2 and Paenibacillaceae did not differ in BJ-FQ2.
Discussion
Research on microorganisms found on the surface of processing tomatoes is essential to help identify microbial communities and characterize regional microbial specialization, aiding disease prevention and quality control efforts (Pinceborde and Casas, 2019). In this study, the fungal and bacterial communities of processing tomato samples from 10 regions were effectively identified by rDNA sequencing to represent the general microbial traits found on processing tomatoes in Xinjiang.
Most (97%) of the OTU classification with sequence identity is equivalent to sequence polymorphism at the species level and can aid microbiological identification (Blaxter et al., 2005). The species accumulation and OTU rank abundance curves in the alpha diversity study demonstrated the validity of the sampling and sequencing techniques.
By searching various databases, the OTUs were organized into several fungal and bacterial taxa. Ascomycota and Basidiomycota were the most prevalent fungal phyla detected on tomatoes, confirming prior research findings on tomatoes (Dong et al., 2020; Zhang et al., 2021), apples (Khwantongyim et al., 2021), and grapes (Bokulich et al., 2014). An average of 20 fungal genera were observed on processing tomatoes. Alternaria had a high abundance in northern Xinjiang, indicating that there was greater plant pathogenic risk in this area.
Pathogenic bacteria, on the one hand, can lead to disease in crops and agricultural products; on the other hand, they can also directly or indirectly enter the human body leading to foodborne diseases. The researchers found total coliform bacteria, yeast, and mold present from fresh tomatoes (Zoellner et al., 2016). Leff and Fierer (2013) demonstrated that fruits and vegetables harbored diverse bacterial communities, related to the type of production, dominated by by taxa belonging to the Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria phyla.
The results of this study were similar to the present study. Pathogens capable of causing human diseases include bacteria, viruses, and parasites that may be present in water used for irrigation or in soil in which produce is grown (Beuchat, 1996). The current study identified comprehensive and detailed microbial communities on processing tomatoes; however, the biological functions of particular species remain unclear. Further study of OTU-based microbial identification is required to fully characterize the tomato microbiome (Magoč and Salzberg, 2011).
There were differences in the microbial communities found on processing tomatoes in southern and northern Xinjiang. Despite extensive work, our understanding of the diversity of microbial communities associated with agricultural products, and the factors that influence the composition of these communities, remains limited (Leff and Fierer, 2013). In our study, tomato varieties grown in different regions may have been important drivers of the fruit microbiome.
The microbial community composition also varies among produce types (Redford et al., 2010). Geographical location, pH, temperature, ammonia, nitrate, total nitrogen, sulfate and hydrogen sulfide, and dissolved organic and inorganic carbon are abiotic factors that influence microbial community composition (Mitrović et al., 2022). Different production types are environmental factors that influence the microbial community composition.
Similarly, differences in growing conditions, transportation procedures, and storage conditions may affect the diversity and composition of microbial communities associated with agricultural products. Farming practices may also have an important, but understudied, influence on the composition of produce-associated microbial communities (Leff and Fierer, 2013). Moreover, different soil properties or ecological characteristics may also be important factors for microbial differences (Pinceborde and Casas, 2019).
The presence of these bacteria on processing tomatoes is also a reflection of the region's environmental characteristics. Xinjiang is primarily dry and rainy with sandy soil, a windy climate, and low vegetation coverage. The susceptibility of processing tomatoes to dust and soil pollution may thus cause the emergence of gut microbes. Southern Xinjiang had a higher abundance of Pantoea, Pectobacterium, and Enterococcus, whereas northern Xinjiang had greater levels of Erwinia, Enterobacter, and Kocuria.
This is similar to the bacterial results isolated by Bah et al. (2019) in the fermented tomato samples. Escherichia coli was detected in two blackberry samples from fresh fruit regions of Italian berries (Macori et al., 2018). Differences in microbial types emphasized the need to prevent orchard diseases, eliminate pathogenic fungi, and conduct post-harvest cleaning. The correlation between environmental factors and microbial specificity should be further researched to confirm this theory. The current study emphasized the microbial differences of processing tomatoes in different regions and provided a basis for further prevention and quality control of processing tomato diseases.
Conclusions
This study investigated the microbial communities on processing tomatoes in two main processing tomato production areas of Xinjiang. Detailed classification of fungi and bacteria was obtained by microbial rDNA sequencing. Fungal diversity was higher in southern than northern Xinjiang. There were also significant differences in fungal and bacterial communities on processing tomatoes in both regions. Out of the 133 different microbial genera identified, significant differences among groups were observed in 11 fungal and 52 bacterial genera.
The differences in the fungi and bacteria on processing tomatoes from two main production areas will provide a basis for the screening of foodborne pathogenic microorganisms in processing tomatoes. Meanwhile, differences in the microbial characteristics of processing tomatoes reflected the environmental specificity of the southern and northern Xinjiang regions, which is of great significance for the prevention and control of regional tomato diseases and biological control strategies. However, the correlation between environmental factors and microbial specificity requires more exploration in our future research.
Footnotes
Acknowledgment
The authors acknowledge the Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China) for their help in DNA sequencing.
Authors' Contributions
S.L., Y.F., and C.W.: methodology, validation, formal analysis, writing-original draft, and writing-review and editing. J.H., F.L., Y.D., X.L., E.Y., S.W., and F.W.: revising and editing. All authors contributed to the article and approved the submitted version.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Key Research and Development Program of China (2019YFC1604502), the agricultural science and technology extension and service project of Xinjiang Uygur Autonomous Region (NTFW-2022-14), and the National Natural Science Foundation of China (32260634).