
Abstract
Vibrio vulnificus is a hazardous foodborne pathogen responsible for approximately 95% of seafood-related deaths. This highlights the urgent requirement for specialized detection tools to be developed and used by food enterprises and food safety authorities. The DETECTR (DNA endonuclease targeted CRISPR trans reporter) system that combines CRISPR/Cas and recombinase polymerase amplification (RPA) has been utilized to develop a molecular detection assay for V. vulnificus. However, because the incompatibility between RPA and Cas12a cleavage has not been addressed, it is a two-step assay that lacks convenience and presents contamination risk. Here, we developed a one-step RPA-CRISPR assay for V. vulnificus using a special crRNA targeting a sequence with a suboptimal protospacer adjacent motif (PAM). The entire assay, conducted at 37°C, takes only 40–60 min, yields results visualized under blue light, and exhibits exceptional specificity and sensitivity (detecting 4 pathogen genome copies per reaction). This study offers a valuable tool for detecting V. vulnificus, aiding in foodborne infection prevention, and exemplifies one-step RPA-CRISPR assays managing Cas-cleavage activity through PAM adjustments.
Introduction
Vibrio vulnificus, responsible for approximately 95% of seafood-related deaths, is an important foodborne pathogen. Seafood contaminated by V. vulnificus can cause wound infections and life-threatening septicemia and acute gastroenteritis (Baker-Austin and Oliver, 2018; Elmahdi et al., 2016; Oliver, 2013). With the globalization of food supply chains nowadays, seafood consumption is growing high, expanding the risk of infection by this dangerous pathogen. This highlights the urgent requirement for specialized detection tools on V. vulnificus to be developed and used by food enterprises and food safety authorities for precise and efficient prevention.
For this purpose, molecular technologies have been introduced for the detection of V. vulnificus. These include polymerase chain reaction (PCR)-based methods that require sophisticated thermal cycling instruments (Bonny et al., 2022; Kim et al., 2008) and isothermal amplification-based methods with easier operations for on-site detection (Han and Ge, 2010; Yang et al., 2020). The use of isothermal technologies, loop-mediated isothermal amplification (LAMP) and recombinase polymerase amplification (RPA), has significantly improved the operational convenience, while the reliability has to be enhanced due to nonspecific amplifications (Han and Ge, 2010; Yang et al., 2021).
Nucleases with sequence-recognition capacity have been combined with isothermal amplifications to enhance the reliability of detection assays (Chen et al., 2018; Feng et al., 2021; Wang et al., 2020). Particularly, Cas12a from the CRISPR/Cas system has had a significant impact on the field of molecular detection (Mao et al., 2022; Yin et al., 2021). Cas12a plays a key role in target recognition by specific binding to the target DNA with the assistance of CRISPR-RNA (crRNA) and inducing cleavage at the target site (Swarts et al., 2017; Zetsche et al., 2015). In addition, Cas12a exhibits nonspecific trans-cleavage activity, enabling it to cleave any single-stranded DNA (ssDNA) present in the reaction mixture (Chen et al., 2018). In the context of detection, the trans-cleavage activity is harnessed to generate a signal when a specially modified ssDNA reporter is introduced to the reaction (Chen et al., 2018; Liang et al., 2019; Wang et al., 2023a). The combination of Cas12a and RPA, known as DETECTR (DNA endonuclease targeted CRISPR trans reporter) (Chen et al., 2018), has been extensively used for molecular detection of various targets, including cancer biomarkers, bacterial pathogens, and viruses (Ding et al., 2020; Huang et al., 2020; Jirawannaporn et al., 2022; Zhong et al., 2022).
There is an inherent limitation in the DETECTR system, the incompatibility between the nucleic acid amplification through RPA and the nuclease cleavage by Cas12a (Chen et al., 2022; Hu et al., 2022; Lin et al., 2022). When RPA and Cas12a are combined in a single reaction, the nuclease activity of Cas12a, stimulated by the target and the amplicons, can potentially degrade the newly synthesized amplicons and primers, thereby restricting the amplification process. This compromises the overall sensitivity of the detection. To retain sensitivity, researchers have often adopted a “two-step” DETECTR strategy in which RPA and Cas12a are separated (Huang et al., 2022; Li et al., 2022; Xiong et al., 2020). However, this approach may sacrifice operational convenience, and there is a risk of cross-contamination during liquid transfer. Numerous efforts have been made to develop “one-step” DETECTR assays. These include integration of the amplification–cleavage “two-step” procedure into one pot (Wang et al., 2019; Zhang et al., 2023), optimization of the reaction components to achieve a balance between amplification and cleavage (Feng et al., 2021; Shao et al., 2023; Wang et al., 2023c; Wang et al., 2023d), use of chemically modified crRNA for controlled activation (Hu et al., 2022), application of multiphase aqueous system for “semiseparation” of the reactions (Lin et al., 2022; Yin et al., 2020), adoption of microfluidic devices (Chen et al., 2020), and adjustment of the cleavage potency by protospacer adjacent motif (PAM) manipulations (Ding et al., 2020; Lin et al., 2023; Lu et al., 2022; Wang et al., 2023b). PAM manipulation has an advantage over the other “one-step” approaches, in that it can tolerate some disruption of the reaction, and thus has stronger adaptability to the on-site detection environment. The other approaches, on the contrary, have to maintain the delicate positioning, concentrations, activator, or aqueous phases during the detection. In addition, microfluidic devices have the cost disadvantage.
For V. vulnificus, a “two-step” DETECTR assay has been developed (Xiao et al., 2021), which needs further improvement to transit to a one-step assay for better convenience and less cross-contamination risk. Here, a one-step RPA-CRISPR assay has been developed utilizing a special crRNA targeting sequence with a suboptimal PAM. The RPA and Cas12a reagents are combined allowing for a one-step assay that is conducted at 37°C for 60 min. The detection results can be observed in real time using a fluorescence detector or at the endpoint using blue light (Fig. 1A). In the one-step reaction, Cas12a exhibits two different cleavage activities upon activation. The cis-cleavage digests the DNA target and the newly synthesized amplicons, while the trans-cleavage cuts the primers and the fluorescence reporter, which is an ssDNA modified with FAM at one end and a quencher at the other (Fig. 1B). However, a highly activated Cas12a may negatively impact the one-step detection assay by causing excessive cleavage, resulting in digestion of target fragments/amplicons and a low fluorescence signal. This issue was encountered when a canonical PAM was selected to design the crRNA. In contrast, when a crRNA designed based on a suboptimal PAM (referred to as scrRNA in this study) was utilized, the Cas12a activation was moderate, leading to less cleavage, more amplicons, and a high fluorescence signal (Fig. 1C). After extensive screening and optimization, a one-step RPA-CRISPR assay using scrRNA was successfully established. This assay demonstrates good specificity and exceptional sensitivity, with the ability to detect 4 copies of the genome per reaction. The entire assay can be completed within 40–60 min at 37°C, with the results visualized under blue light. The one-step reaction achieves a well-balanced amplification by RPA and digestion by Cas12a, serving as a valuable example for the development of one-step RPA-CRISPR assays. In addition, this study provides a useful tool for molecular detection of V. vulnificus, contributing to the prevention of serious foodborne infections.

Illustration of the one-step RPA–CRISPR assay.
Materials and Methods
Bacteria
Bacterial strains used in this study include V. vulnificus, V. cholerae, V. parahaemolyticus, V. mimicus, V. alginolyticus, V. shilonii, V. ichthyoenteri, V. splendidus, Yersinia enterocolitica, and Staphylococcus aureus (identification numbers: ATCC 27562, ATCC 14100, ATCC 17802, MCCC 1A02602, ATCC 17749, ATCC BAA-91, MCCC 1A00057, MCCC 1A04096, CMCC 52204, and ATCC 6538, respectively). These strains were subjected to 16S rRNA sequencing. DNA extraction was performed with a Tiangen DNA Kit, Beijing, China. Quantification of DNA was done with a NanoDrop, Thermo Fisher, Wilmington, USA. The target copy number (cps) was calculated using the genome size (Yang et al., 2020).
Preparation of Cas12a
The Cas12a protein, encoded by the LbCpf1 gene (GenBank no. OK557998.1), was synthesized within the context of pET-28b(+) to create an expression plasmid with an N-terminal His-Tag (Fig. S1A). The accuracy of the plasmid was verified by DNA sequencing, General Biology, Anhui, China. Cas12a was induced using 0.5 mM (final) of IPTG in E. coli BL21-pLysS at 23°C for 8 h. The overexpressed Cas12a was purified by the Sangon Biotech Ni-affinity resin, Shanghai, China, on a AKTA Prime Plus, GE Healthcare Life Sciences, and stored at −20°C in 500 mM NaCl, 1 mM TCEP, 25 mM Tris-HCl, pH 7.5, and 50% glycerol. The expression and purification of Cas12a were analyzed using SDS-PAGE (Fig. S1B). To confirm the purified Cas12a was free of nonspecific nuclease activity, 50 nM (final) of the purified Cas12a was incubated with 150 ng of pUC18 plasmid in 20 μL of 1× NEB buffer 2.1 at 37°C for 1 h and analyzed by agarose gel electrophoresis (Fig. S1C).
crRNA preparation
The empV gene (GenBank no. U50548.1, gene product extracellular metalloproteinase), which had been established as a valid target for molecular detection of V. vulnificus (Yang et al., 2020), was used in this study as the detection target. A previously established DNA fragment on this gene was used as the basis for designing crRNAs, as shown in Table 1 and Figure S2 (Wang et al., 2021; Yang et al., 2021). A BLAST search of this particular fragment returned only the V. vulnificus sequences, further confirming the species specificity of this fragment. The crRNAs were prepared by an in vitro transcription. DNA substrates containing T7 promoter and crRNA sequences were generated by annealing two reverse-complement single-stranded oligonucleotides (10 μM each, sequences provided in Table S1) at room temperature following a denaturation step at 95°C for 5 min. After quantification using NanoDrop (Thermo Fisher), 1 μg of the DNA template was transcribed at 37°C for 16 h using T7 transcription (Vazyme Biotech, Nanjing, China). Transcription products were treated by DNase I, purified, quantified using Qubit 4 (Thermo Fisher), and stored at −80°C.

Canonical PAM could facilitate two-step RPA–CRISPR.
Oligonucleotide Sequences
Underlined sequences are complementary to the target fragment. RPA, recombinase polymerase amplification; qPCR, quantitative PCR.
RPA reaction
A lyophilized enzyme pellet of the RAA Basic Reagent, Hangzhou ZC Bio-Sci & Tech, Zhejiang, China, was reconstituted by 25 μL of buffer A, 19.25 μL of ultrapure water, and 1.5 μL each of the primers (20 μM). This RPA mixture (15.75 μL) was combined with 1.25 μL of buffer B and 3 μL of template DNA, and incubated at 37°C for 30 min. The RPA products were analyzed using agarose gel electrophoresis.
CRISPR reaction
The CRISPR reaction of 20 μL comprised 50 nM of crRNA, 50 nM of Cas12a, 500 nM of ssDNA-FQ, and 2 μL of the RPA amplicon in 1× NEB buffer 2.1. The reaction was conducted for 60 min under 37°C on a Roche Light Cycler 480 II qPCR system with FAM detection (465–510 nm). The endpoint signal was observed with a blue light of the Novogene transilluminator, Tianjin, China. The endpoint images were taken by a smartphone camera. Quantification of the fluorescence signal from the images was done with the ImageJ intensity analysis software (National Institutes of Health, MD, USA).
One-step RPA-CRISPR
For the RPA premix, the lyophilized enzyme pellet of the RAA basic reagent was reconstituted with 25 μL of buffer A and 1.5 μL of each primer (20 μM). For the CRISPR premix, 250 nM of crRNA, 125 nM of Cas12a, and 1.25 μM of the ssDNA-FQ were mixed in 2.5× NEB buffer 2.1. A total of8 μL of CRISPR premix, 7.75 μL of RPA premix, 3 μL of template DNA, and 1.25 μL of buffer B constituted the one-pot reaction. The one-step assay was performed for 60 min at 37°C using a Roche Light Cycler 480 II qPCR system with FAM detection (465–510 nm). The endpoint signal was visualized under blue light (Novogene transilluminator). The camera images were analyzed with the ImageJ software.
Preparation of the spiked samples
Fresh oysters were bought from a local seafood market in Lianyungang, China, and verified to be free of V. vulnificus using quantitative PCR (qPCR) (D’Souza et al., 2019). The oyster tissue was homogenized using a handheld grinder. Negative samples were created by combining 1 g of homogenate with 10 mL of alkaline peptone. For positively spiked samples, 1 g of homogenate was mixed with 10 mL of V. vulnificus alkaline peptone culture with the desired bacterial density. For the validation with randomly spiked samples, a total of six positively spiked samples and four negative samples were prepared and randomly numbered for testing. DNA was extracted from 300 μL of the sample with a Tiangen DNA Kit. The randomly spiked samples had a loading amount of 101–103 colony forming units (CFUs) per reaction. One CFU was considered to have 1 copy of the target because there was one copy of the empV gene per genome.
qPCR
The qPCR procedure followed the methodology described in a previous study (D’Souza et al., 2019). Positive results were determined based on cycle threshold (Ct) values below 32.
Results and Discussion
Canonical PAM could facilitate two-step RPA-CRISPR
By utilizing the RPA primers and the crRNA sequence (crRNA3) from a previously reported assay (Table 1) (Zhang et al., 2023), we achieved a high sensitivity of 4 copies of the target gene per reaction using the “amplification–cleavage” two-step procedure (Fig. 2A). This indicated that the canonical PAM could facilitate two-step RPA-CRISPR. The RPA was efficient when separated from the CRISPR reaction. Although not as distinguishable as the fluorescence signal, the amplification product was detectable through gel analysis with as few as 4 copies of the target (Fig. 2B). However, when we directly mixed these RPA and CRISPR components for a one-step reaction, the sensitivity was significantly compromised(Fig. 2C). The sensitivity dropped sharply to only 4 × 104 copies/reaction compared with the two-step procedure, despite using the same materials. This clearly indicated an incompatibility between the RPA and the CRISPR cleavage in this V. vulnificus detection system, where the excessively activated Cas12a digested the target fragments and the new amplicons, as we had speculated in Figure 1.
Establishing a one-step RPA-CRISPR assay using scrRNA
We used the PAM manipulation approach to establish the one-step RPA-CRISPR assay for V. vulnificus detection, which involved screening of four suboptimal PAMs in the RPA amplicon sequence that complied with the pattern of VTTV or TCTV (‘V’ refers to A, C, or G) (Fig. S2) (Lu et al., 2022; Yamano et al., 2017). Based on these suboptimal PAMs, four corresponding scrRNAs (scrRNA1-4) were designed (Table 1). Through a two-step procedure, all four scrRNAs were confirmed to possess the fundamental functionality of activating Cas12a (Fig. 3A). Subsequently, these scrRNAs were evaluated in a one-step assay. The endpoint signals and fluorescence curves demonstrated their effectiveness in the one-step assay, with scrRNA2 exhibiting the highest signal intensity (Fig. 3B). As anticipated, crRNA3 only functioned in the two-step procedure but not in the one-step assay. The suboptimal scrRNA2 was selected for the one-step RPA-CRISPR assay, and its concentration was determined to be at least 100 nM (Fig. 3C).
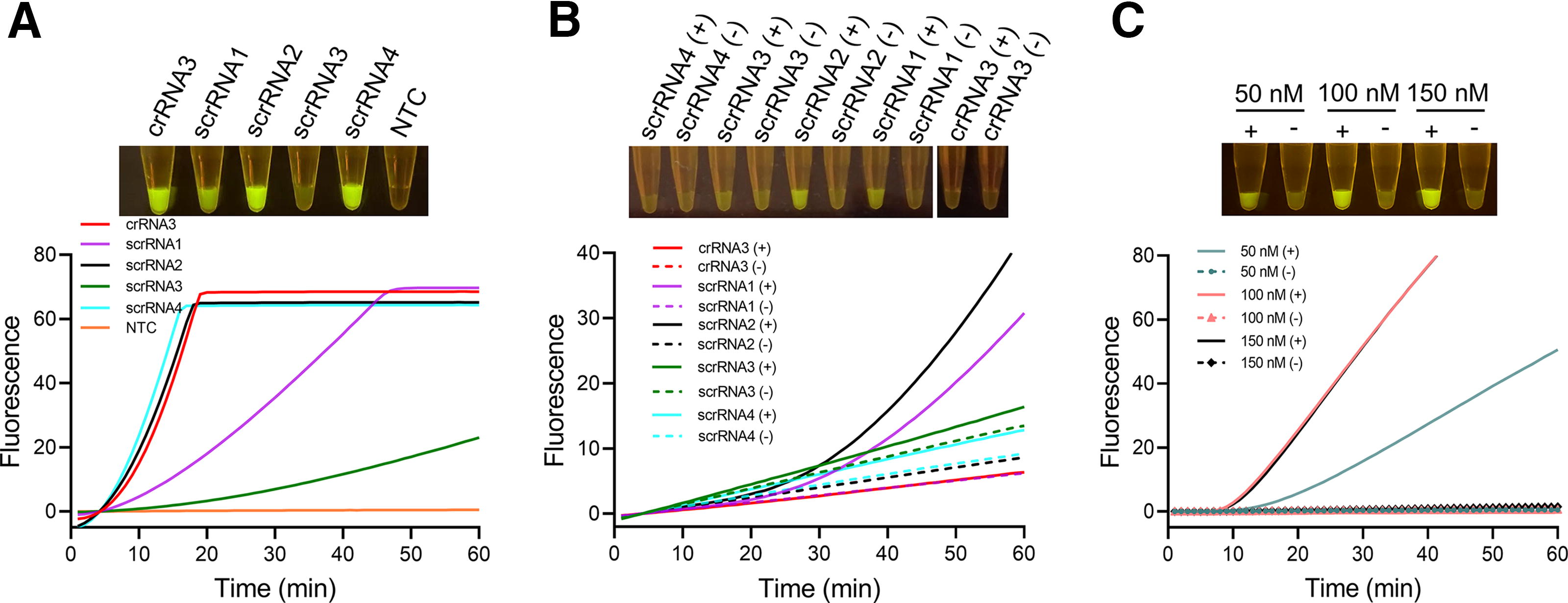
Establishing a one-step RPA–CRISPR assay using scrRNA.
Evaluation of the one-step assay
The sensitivity was evaluated by using different amounts of V. vulnificus genomic DNA, ranging from 4 × 10° copies to 4 × 105 copies. Both the fluorescence curve and the endpoint signal results demonstrated a sensitivity of 4 copies per reaction, which was comparable with the two-step assay (Fig. 4A–C) (Xiao et al., 2021). When compared with other molecular assays reported in the literature, such as RPA (2 copies/reaction) LAMP (1–10 copies/reaction), and qPCR (∼15 copies/reaction), this sensitivity was not inferior (D’Souza et al., 2019; Han and Ge, 2010; Yang et al., 2021). The fluorescence curves also indicated that 40 min was sufficient to generate a clear signal.

Sensitivity and specificity of the one-step RPA–CRISPR assay.
The specificity was assessed using various Vibrio species and two other foodborne pathogens, namely V. mimicus, V. cholerae, V. alginolyticus, V. shilonii, V. ichthyoenteri, V. splendidus, V. parahaemolyticus, S. aureus, and Y. enterocolitica. The results demonstrated the excellent specificity of the assay, with only V. vulnificus generating a detectable signal (Fig. 4D). The target gene (empV) had already been established in previous studies (Wang et al., 2021), showing reliable specificity and evolutionary conservation. The target fragment used in this study was species-specific, and an alignment of the target fragment sequences from 13 V. vulnificus GenBank inputs (all returned from a BLAST search of the RPA amplicon sequence) suggested high conservation of the scrRNA2 corresponding area (Fig. S3). The selection of this target fragment in gene empV and the design of scrRNA2 ensured the specificity of the assay.
Validation with spiked samples
The validation of the one-step RPA-CRISPR assay involved a sensitivity test of the assay using the quantitatively spiked samples, and detection of V. vulnificus in 10 randomly spiked samples (S1–S10). The results of the sensitivity test showed that the assay could detect as low as 4 CFUs of V. vulnificus in the spiked samples (Fig. 5A), which was equivalent to the 4 target copies as above. For the detection of randomly spiked samples, the outcomes were compared with those obtained from the two-step RPA-CRISPR assay and qPCR (Fig. 5B, S4 and Table S2). Consistent with the preparation of the randomly spiked samples, the test detected six positive samples and four negative samples. Moreover, the results of the one-step assay were completely consistent with those of the two-step assay and qPCR.
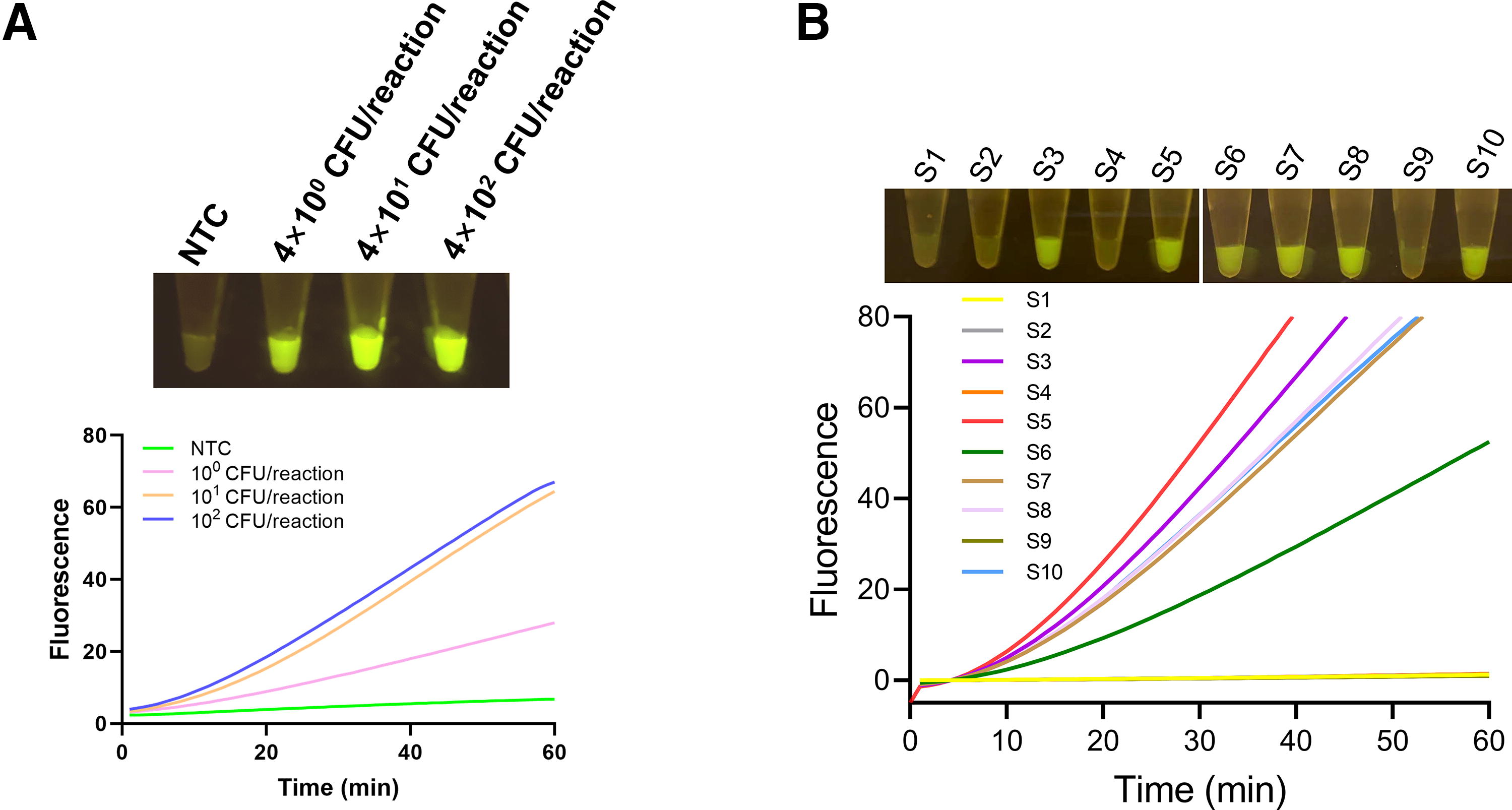
Validation of the one-step RPA–CRISPR assay with spiked samples.
Technological contributions
The PAM is a significant factor in CRISPR/Cas12a-mediated nucleic acid cleavages. From the molecular detection perspective, the requirement of PAM limits the flexibility of assay development on target sequence selection, and meanwhile provides a tool to manage the cleavage activity of Cas12a for certain applications. With the urgent demand for sensitive and reliable detection tools for SARS-CoV-2 in the past few years, Cas12a-based assays that utilized PAM manipulations have been developed, including the use of suboptimal PAM (Lu et al., 2022) and selecting sequences without the PAM (Ding et al., 2020; Lin et al., 2023). Another pathogen detection report for acute hepatopancreatic necrosis disease has applied suboptimal PAM to facilitate one-step RPA-CRISPR (Wang et al., 2023b). These assays have detection sensitivities ranging from several copies to hundreds of copies according to the detection requirements, suggesting that the PAM manipulation can keep the assay sensitivity if optimized appropriately. To date, there are only limited numbers of literatures dealing with the PAM to solve the incompatibility of Cas12a. Developing the one-pot system using PAM manipulation has certain advantages. The approaches using compartmentation of reaction reagents in one pot have a higher requirement on the operate procedure because accidental mixing of the reagents with wrong timing can disrupt the reactions. The approaches controlling the concentrations of reaction components in one pot have more pressure on the reagent manufacture and transportation because small inaccuracies of the component concentrations can affect the detection results. Therefore, a one-pot system using appropriate PAM manipulation is more robust, and exploring the general rules for PAM manipulation has a good value for the utilization of CRISPR/Cas. This study offers another example that helps establish the general principle of managing Cas-cleavage activity through PAM adjustments.
Evaluations of the one-step RPA-CRISPR assay for V. vulnificus detection show that it maintains the same level of accuracy and reliability as the two-step assay, while offering improved convenience in operation and better control over carryover contaminations. The assay can be completed within 40–60 min under isothermal conditions, and the results can be visually interpreted. The excitation wavelength for FAM is 494 nm, which is within the wavelength range of the blue light (Wang et al., 2023d). Thus, visualization of the detection results does not require a UV source. A blue light is good enough to visualize the results for the photograph taken and software analysis. The assay can be easily used for on-site detection of V. vulnificus.
Conclusions
This research presents a one-step RPA-CRISPR method for detecting V. vulnificus, enabling simultaneous amplification of nucleic acids and Cas12a cleavage. The issue of incompatibility between RPA and CRISPR cleavage has been resolved by using a specific crRNA targeting a sequence with a suboptimal PAM. This one-step assay demonstrates excellent specificity and high sensitivity, capable of detecting as few as 4 genomic DNA copies. The entire process is carried out at 37°C within 40–60 min with a visualized outcome reading under blue light. This study serves as a commendable model for developing one-step RPA-CRISPR assays and offers a valuable tool for molecular detection of V. vulnificus, contributing to the prevention of severe foodborne infections. Establishment of an integrated device that includes sample preparation, signal visualization, and result interpretation can be one of the future directions.
Footnotes
Authorship Contribution Statement
X.Z.: Conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing—original draft, and writing—review and editing. Y.W.: Conceptualization, data curation, methodology, investigation, validation, writing—original draft, and writing—review and editing. Y.T.: Methodology, investigation, and validation. L.Y.: Conceptualization, methodology, and investigation. C.Z.: Methodology and investigation. G.Y.: Conceptualization, data curation, formal analysis, project administration, visualization, supervision, writing—original draft, and writing—review and editing. P.W.: Conceptualization, data curation, formal analysis, investigation, project administration, visualization, supervision, writing—original draft, writing—review and editing, and funding acquisition. S.G.: Conceptualization, data curation, formal analysis, project administration, supervision, resources, writing—original draft, writing—review and editing, and funding acquisition.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants from the China Postdoctoral Science Foundation (2022M721665), the Jiangsu Funding Program for Excellent Postdoctoral Talent of China (2022ZB358), and the Priority Academic Program Development of Jiangsu Higher Education Institutions of China.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.