
Abstract
Listeria monocytogenes is a critical foodborne pathogen that causes severe invasive and noninvasive diseases and is associated with high mortality. Information on the prevalence of L. monocytogenes infections in Taiwan is very limited. This study aimed to analyze the molecular epidemiological surveillance and virulence gene distribution of 176 human clinical L. monocytogenes isolates collected between 2009 and 2019 in northern Taiwan. Our results showed that the isolates belonged to 4 serogroups (IIa, IIb, IVb, and IIc), with most isolates in serogroups IIa (81/176, 46%) and IIb (71/176, 40.3%). Multilocus sequence typing analysis revealed 18 sequence types (STs) and 13 clonal complexes (CCs). Eighty-four percent of all isolates belonged to six STs: CC87-ST87 (40/176, 22.7%), CC19-ST378 (36/176, 19.9%), CC155-ST155 (28/176, 15.5%), CC1-ST710 (16/176, 8.8%), CC5-ST5 (16/176, 8.8%), and CC101-ST101 (11/176, 6.1%). Furthermore, our analysis showed the distributions of four Listeria pathogenicity islands (LIPI) among all isolates. LIPI-1 and LIPI-2 existed in all isolates, whereas LIPI-3 and LIPI-4 only existed in specific STs and CCs. LIPI-3 existed in the STs, CC1-ST710, CC3-ST3, CC288-ST295, and CC191-ST1458, whereas LIPI-4 could be found in the STs, CC87-ST87 and CC87-ST1459. Strains containing LIPI-3 and LIPI-4 are potentially hypervirulent; thus, 68/176 isolates (39.1%) collected in this study were potentially hypervirulent. Since L. monocytogenes infections are considered highly correlated with diet, molecular epidemiological surveillance of Listeria in food is important; continued surveillance will provide critical information to prevent foodborne diseases.
Introduction
L
Several molecular epidemiological surveillance methods have been developed for L. monocytogenes, including serotyping, geno-serotyping (multiplex polymerase chain reaction [PCR]-serotyping) (Doumith et al., 2004), pulsed-field gel electrophoresis (PFGE) (Wang et al., 2015), and multilocus sequence typing (MLST) (Salcedo et al., 2003). To date, 14 serotypes (1/2a, 1/2b, 1/2c, 3a, 3b, 3c, 4a, 4b, 4c, 4d, 4e, 4ab, 4h, and 7) have been discovered to classify L. monocytogenes (Feng et al., 2020; Yin et al., 2019), of which 4 serotypes (1/2a, 1/2b, 1/2c, and 4b) comprise the major pathogenic groups. Among these four types, serotypes 1/2a, 1/2b, and 4b are considered responsible for the most frequent listeriosis outbreaks (Bouymajane et al., 2021; Maung et al., 2023). Twenty-two clonal complexes (CCs) and 4 evolutionary lineages were created for L. monocytogenes using serotypes and MLST (Ragon et al., 2008), where 80% of the strains belonged to 12 CCs and 3 evolutionary lineages. One of the evolutionary lineages (lineage I) contained the most virulent strain of L. monocytogenes (Maury et al., 2016).
Listeriosis depends on different virulence factors that may be responsible for different stages of the infection process (Dussurget et al., 2014). These virulence factors were found to belong to four different pathogenicity islands (Listeria pathogenicity islands [LIPI]-1 to LIPI-4). LIPI-1 and LIPI-2 were found in most isolates (Chen et al., 2020; Zhang et al., 2019b). LIPI-1 contains six important virulence genes (hly, prfA, plcA, plcB, mpl, and actA), which are responsible for intracellular parasitism (Hadjilouka et al., 2018; Poimenidou et al., 2018). LIPI-2 contains the inlAB operon that encodes two internalins, InlA and InlB, which mediate host cell invasion (Cossart et al., 2003; McGann et al., 2007). LIPI-3 was only found in strains belonging to lineage I (Quereda et al., 2016), and encodes listeriolysin S, which has both hemolytic and bactericidal activity to help strain survival during infection (Cotter et al., 2008; Quereda et al., 2017). LIPI-4 is the last identified pathogenic island and is primarily identified in CC4 strains. It encodes the cellobiose-family phosphotransferase system (PTS) and is potentially involved in placental tropism infection (Maury et al., 2016).
Although each LIPI contains various virulence genes, LIPI-3 and LIPI-4 are considered more hypervirulent than the other two (Zhang et al., 2020). Since LIPI-3 and LIPI-4 are associated with L. monocytogenes lineages and sequence types (STs) (Zhang et al., 2019b), monitoring the evolution of hypervirulent populations can be efficiently achieved by surveilling LIPI-3 and LIPI-4.
Owing to the lack of prevalence studies, little is known about the prevalence of L. monocytogenes in Taiwan. During a food sample investigation in 2012, data showed a nearly 5.5% distribution of L. monocytogenes in Taiwanese food (Wang et al., 2012) such as meat, its related products, and vegetables; however, this investigation contained no molecular typing information for further analysis. Recently, a comprehensive nationwide surveillance in Taiwan showed that both CC87 and CC378 accounted for the predominant groups in food and clinical cases (Tsai et al., 2022), providing an updated prevalence information. To understand whether a difference between the local and nationwide prevalence exists, we conducted surveillance using various molecular typing and PCR methods to detect whether specific clonal lineages and molecular types of clinically isolated L. monocytogenes with virulence genes existed in northern Taiwan.
Materials and Methods
Bacterial strains
A total of 176 human, clinically isolated L. monocytogenes strains (148 from blood, 20 from body fluids, 5 from wounds, 2 from urine, and 1 from sputum) were confirmed using Matrix-assisted laser desorption/ionization, time of flight at Chang Gung Memorial Hospital, Linkou in northern Taiwan from 2009 to 2019. All isolates were stored in tryptic soy broth containing 20% glycerol at
Strain Origin and Year of Isolation
B, blood; BF, body fluid; CSF, cerebrospinal fluid; SP, sputum; TS, tissue or pus; U, urine.
PCR serogroup analysis
PCR serogroup analysis was performed using a multiplex PCR assay as previously described (Doumith et al., 2004; Kerouanton et al., 2010). DNA was extracted using the QIAamp DNA Kit (QIAGEN®). The target genes for discriminating different serogroups included prs, lmo737, lmo1118, ORF2110, and ORF2819. The primers used for multiplex PCR are listed in Table 2. Five serogroups (IIa, IIb, IIc, IVa, and IVb) were identified through the different PCR products; serogroup IIa (serotypes 1/2a and 3a) was confirmed using lmo737, serogroup IIb (serotypes 1/2b and 3b) was confirmed using ORF2819, serogroup IIc (serotypes 1/2c and 3c) was confirmed using both lmo737 and lmo1118, serogroup IVa (serotype 4c) was confirmed using prs, and serogroup IVb (serotypes 4b and 4ab) was confirmed using both ORF2819 and ORF2110. The PCR was performed as follows: initial DNA denaturation at 94°C for 3 min; 35 cycles of amplification followed by denaturation at 94°C for 24 s, 53°C for 69 s, and 72°C for 69 s; and final extension at 72°C for 7 min. The PCR products were examined using electrophoresis in 2% agarose gel.
Primers Used for Listeria monocytogenes Serotyping
All PCR conditions were as reported in Doumith et al. (2004).
PCR, polymerase chain reaction.
Multilocus sequence typing
MLST was performed by analyzing seven housekeeping gene sequences, as previously described (Cantinelli et al., 2013; Ragon et al., 2008). The evolutionary and phylogenetic analysis of each STs was performed by eBURST (https://www.phyloviz.net/goeburst).
LIPI virulence gene analysis
Four LIPI virulence genes were examined by PCR, as previously described (Zhang et al., 2019b). The primers used for PCR for LIPI gene analysis are listed in Table 3. The PCR mixtures (25 μL) included 12.5 μL Taq PCR Master Mix (TaKaRa®), 1 μL forward primer, 1 μL reverse primer, and 1.5 μL DNA template. PCR was performed as follows: initial DNA denaturation at 95°C for 2 min; 30 cycles of amplification followed by denaturation at 95°C for 30 s, annealing for 30 s, and elongation at 72°C for 90 s. Primer annealing temperature varied based on the melting temperature of each primer. The PCR products were analyzed by electrophoresis on a 1% agarose gel.
Primers Used to Amplify Listeria monocytogenes Virulence Genes
All PCR conditions were as reported in Zhang et al. (2019b).
LIPI, Listeria pathogenicity islands.
Results
Molecular typing
A total of 18 STs and 13 CCs were identified among the 176 isolates (Fig. 1 and Table 4), and no correlation was observed between the specimen sources and CCs or serotypes (Table 4). The results showed that ST87, ST378, and ST5 were continually found in our collection since 2009; ST87 showed an increasing trend, while ST378, considered an endemic population, showed a decreasing trend (Fig. 1). Our data showed that 147 (84%) isolates belonged to 6 major STs: ST87-CC87 (40/176, 22.7%), ST378-CC19 (36/176, 19.9%), ST155-CC155 (28/176, 15.5%), ST710-CC1 (16/176, 8.8%), ST5-CC5 (16/176, 8.8%), and ST101-CC101 (11/176, 6.1%) (Table 4). In addition to these 6 major STs, strains belonging to other STs did not exceed 10 isolates.

MLST analysis of Listeria monocytogenes distribution from 2009 to 2019. MLST, multilocus sequence typing.
Distribution of Sequence Types, Clonal Complexes, and Virulence Factors of Listeria monocytogenes from 176 Clinical Isolates
ST, sequence type.
The four serogroups, IIa, IIb, IVb, and IIC, are shown in Figure 2 and Table 4. Most isolates belonged to serogroup IIa (82/176, 46.6%), followed by serogroups IIb (70/176, 39.8%) and IVb (22/176, 12.5%). Only two isolates (1.1%) belonged to serogroup IIc. Although most isolates belonged to serogroup IIa, after 2017, serogroup IIb showed an increasing prevalence, and serogroups IIa and IVb showed a decreasing prevalence, whereas no serogroup IVb strains were found. Based on the MLST analysis results, 92 isolates (52.8%) belonged to lineage I, and 82 belonged to lineage II (47.1%), whereas the lineage could not be determined for 2 isolates since they were of new STs. Serogroup IIb was the most abundant serogroup in lineage I and included CC2, CC3, CC5, CC87, CC288, CC1, and CC191. The most abundant serogroup in lineage II was IIa, including CC9, CC7, CC101, CC155, CC177, and CC19. The data showed that various STs had serogroup specificities, except for ST378. Among the 36 ST378 isolates, 35 belonged to serogroup IIa, and 1 belonged to serogroup IIb (Fig. 3).
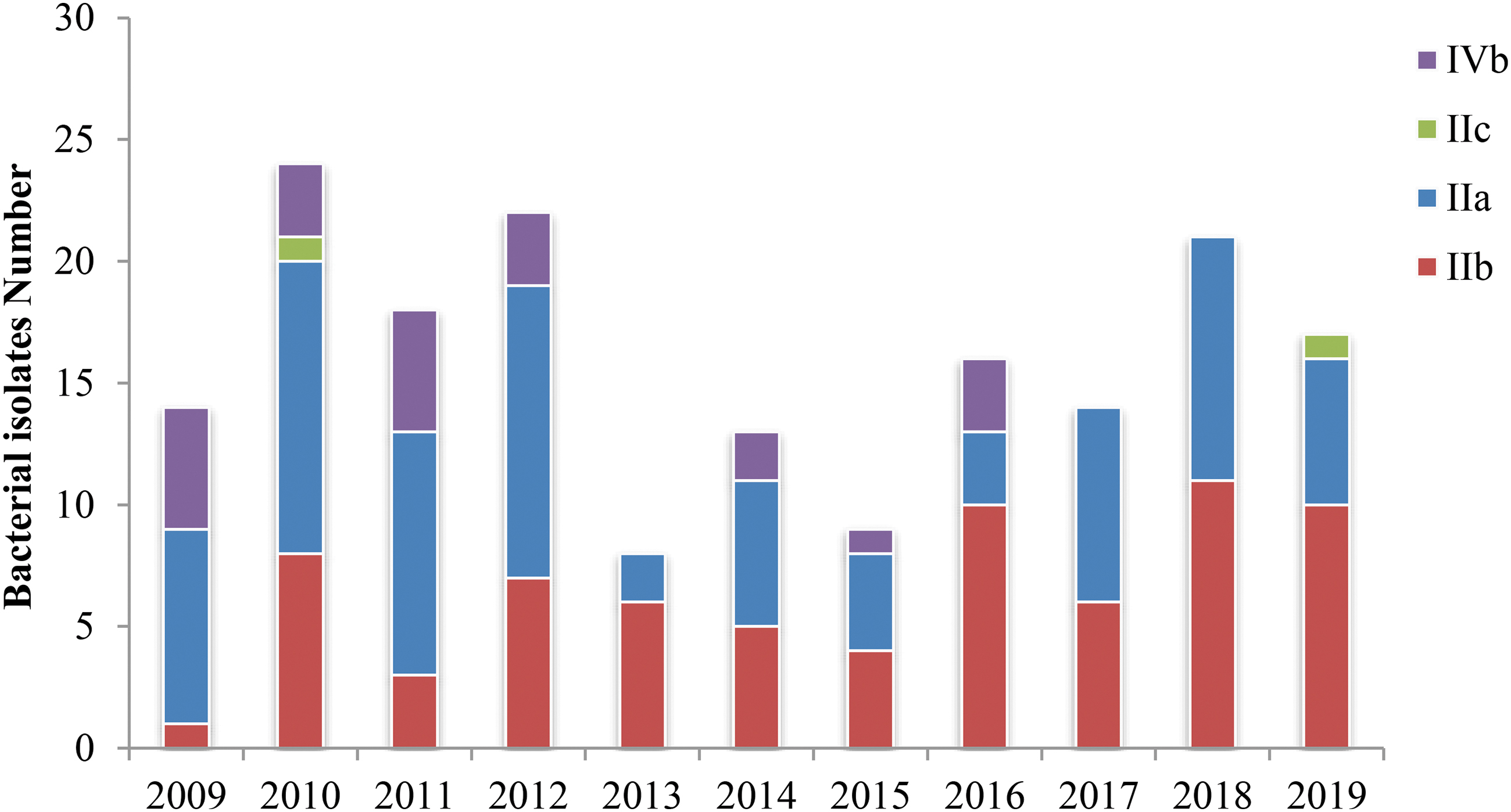
Serogroup distribution of Listeria monocytogenes from 2009 to 2019.

Correlation between MLST-derived lineage and serotype in the clinical Listeria monocytogenes isolates collected in this study.
Virulence gene analysis
Analysis of virulence genes encoded by various LIPIs revealed that 10 virulence genes encoded by LIPI-1 and LIPI-2 were distributed in all isolates, and the genes encoded by LIPI-3 and LIPI-4 correlated with the strain STs and CCs (Table 4). Among the 176 isolates, 27 and 41 contained virulence genes encoded by LIPI-3 and LIPI-4, respectively. All these isolates belonged to lineage I. In addition to lineage specificity, strains containing virulence genes encoded by LIPI-3 belonged to CC3-ST3, CC288-ST295, CC1-ST710, and CC191-ST1458. In contrast, strains containing virulence genes encoded by LIPI-4 were only found in CC87 (ST87 and ST1459). Except 16 CC1-ST710 belonging to serogroup IVb, most of the 68 isolates belonged to serogroup IIb.
Discussion
Our results showed that serogroup IIa was the most prevalent (46%), followed by IIb, which has continued to increase recently. Most serogroup IIb isolates belonged to CC-3 or CC-87, which contain the virulence factors encoded by LIPI-3 or LIPI-4, respectively; these factors are considered responsible for this increasing prevalence. MLST analysis revealed that ST87-CC87, ST378-CC19, ST155-CC155, ST710-CC1, and ST5-CC5 were the major groups, consistent with the findings of previous investigations of L. monocytogenes originating from food in Taiwan (Huang et al., 2015; Tsai et al., 2022) and similar to that reported in China (Zhang et al., 2019a). A clinical listeriosis analysis in China showed that over 92% of the isolates belonged to serotypes 1/2a, 3a, 1/2b, and 3b, and the most abundant STs were ST8, ST5, ST87, and ST155 (Zhang et al., 2019a). A similar trend was also found in L. monocytogenes isolated from food: 1/2a, 1/2b, 1/2c, and 4b were the major serotypes, and the most abundant STs were ST8, ST9, ST155, and ST87 (Lopez-Valladares et al., 2018; Wang et al., 2018; Zhang et al., 2019b).
Aside from those found in Taiwan and China, other regions have reported different trends in prevalence. The major serotype in the central California coast area in the United States and in Europe is 4b (Gorski et al., 2022; Nuesch-Inderbinen et al., 2021). The major serotypes in Austria are 4b, 4e, and 4ab (Jennison et al., 2017). The variations in serotype distribution in different areas may be attributed to various dietary habits (Lopez-Valladares et al., 2018), and further investigations are needed to validate this hypothesis.
Except for two outbreaks reported in Spain, ST87 has rarely been isolated from patients infected with L. monocytogenes in Western countries (Perez-Trallero et al., 2014). In contrast, ST87 has been frequently isolated in patients infected with L. monocytogenes in Taiwan and China (Huang et al., 2015; Zhang et al., 2019a); it is one of the most common STs (22.7%) in our analysis. All our ST87 isolates contained LIPI-4, which was previously considered responsible for central nervous system (CNS) and maternal/neonatal (MN) infections (Maury et al., 2016).
We found that each ST in our collection contained LIPI-1- and LIPI-2-related genes, consistent with a previous report in China (Zhang et al., 2019b). Since the geographic location of Taiwan is close to China, this may have caused a similar strain virulence distribution (Zhang et al., 2019b). In addition to LIPI-1 and LIPI-2, our results showed that LIPI-3 and LIPI-4 have lineage and CC specificities, consistent with a previous report (Maury et al., 2016). Our results demonstrated that LIPI-3 were found most in CC1-ST710, CC3-ST3, CC288-ST295, and CC191-ST1458, all of which belong to lineage I. Except CC1-ST710, the results were consistent with those of previous reports (Clayton et al., 2014; Quereda et al., 2016). CC1-ST710 was rarely found in previous reports, yet 8.8% of the isolates in our study belonged to this CC. Several prevalence data in our investigation were consistent with a recent nationwide surveillance in Taiwan (Tsai et al., 2022); however, CC1-ST710 was not found in this surveillance report either.
CC1-ST710 was only found in isolates from before 2016 in our surveillance, possibly correlating with changes in bacterial communities influenced by several factors such as climate change (Lennon et al., 2023) or antibiotic usage (Nel Van Zyl et al., 2022).
Our survey found that LIPI-4 only exists in CC87, including ST87 and ST1459, aligning with previous studies showing that only LIPI-4 exists in CC4, CC87, and ST619, which are considered responsible for CNS and MN infections (Maury et al., 2016; Wang et al., 2018). LIPI-4-containing strains were potentially hypervirulent (Maury et al., 2016), possibly contributing to CC87 emerging as the major CC (23.6%) in our collection.
Conclusions
Our molecular epidemiological surveillance and virulence gene investigation of clinical Listeria isolates during 2009–2019 yielded several conclusions. Half of the isolates belonged to lineage I, which had a higher virulence. Most of the isolates collected in our study belonged to serogroup IIa, and ST87 was the predominant ST group. Surveillance for hypervirulent LIPI-3 and LIPI-4 confirmed that their distribution has CC specificities, and they could only be found in CC87-ST87, CC87-ST1459, CC3-ST3, CC1-ST710, CC288-ST295, and CC191-ST1458. Although our data and the nationwide surveillance both showed the same predominant ST group, other clonal lineage differences were also found in both surveillance data, which emphasized the necessity of our local study. Since listeriosis is considered highly correlated with consuming contaminated foods (Wang et al., 2012), our analysis showed that molecular epidemiological surveillance of food substances might provide critical information to prevent listeriosis outbreaks.
Footnotes
Acknowledgment
The authors are grateful for the bacterial isolates provided by the Chang Gung Memorial Hospital, Linkou bacterial storage bank program (CLRPG3E0025).
Authors' Contributions
All authors have participated in the conception and design, data analysis and interpretation, drafting, revising, and approval of the final version this article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants from the Chang Gung Memorial Hospital, Linkou, Taiwan (CMRPG3K0371) and the Ministry of Science and Technology, Taiwan (MOST 110-2320-B-182A-006-MY3 and NSTC 112-2811-B-182A-018).