
Abstract
The infection status and etiological analysis of Aeromonas spp. from foodborne diarrhea patients in Wenzhou were carried out to provide the etiological basis for healthy diet and clinical treatment. Aeromonas isolates (n = 41) collected from foodborne diarrhea patients were identified using the automatic bacteriologic analyzer and mass spectrometer. Species identification, multilocus sequence typing, prediction of virulence genes, and antimicrobial resistance genes were analyzed by the data of whole genome sequencing. The antibiotic resistance of these isolates was determined using miniaturization of the broth dilution susceptibility test. A total of 1829 stool samples of diarrhea patients were collected, and the detection rate of Aeromonas spp. was 2.24% (41/1829). Moreover, Aeromonas spp. are more easily detected in warmer months (from June to August), which were identified as follows: A. veronii (53.66%, 22/41), A. caviae (21.95%, 9/41), A. hydrophila (9.76%, 4/41), A. dhakensis (4.88%, 2/41), A. rivipollensis (4.88%, 2/41), A. enteropelogenes (2.44%, 1/41), and A. media (2.44%, 1/41). All strains can be divided into 38 sequence types, 31 of which were novel, suggesting that Aeromonas spp. had high genetic diversity, multiple clones, and various sources in diarrhea patients. High number of genetic diversity and resistance were found in the Aeromonas isolates. In addition, the category distribution of the virulence genes was significantly different among the seven species of Aeromonas. Aeromonas spp. had different degrees of resistance to antibiotics, and tetracycline was the most serious, with a resistance rate of 27%. What’s more, for some antimicrobial classes in silico antimicrobial resistance gene detection was highly correlated with phenotypic antimicrobial resistance patterns with an overall sensitivity of 93.3% and a specificity of 66.7%. The findings from this research highlighted the importance for development of prevention and control strategies to reduce the risk of foodborne diarrhea caused by Aeromonas spp.
Introduction
Aeromonas spp. are a type of Gram-negative brevibacterium with blunt ends, unipolar flagellates, and oxidase positive. Aeromonas spp. are a potential zoonotic pathogen and a major fish pathogen (Awan et al., 2018), which can be found in the natural environment, animal, and human (Bhowmick and Bhattacharjee, 2018). Aeromonas genus comprised 36 species isolated from foods, animals, and various infectious processes in humans, and at least 19 species were considered emerging pathogens to humans which caused a broad spectrum of infections, mainly gastroenteritis, wound infections, and bacteremia/septicemia, infecting immunocompromised and immunocompetent (Fernández-Bravo and Figueras, 2020). In addition, studies have so far reported that 95.4% of the strains associated with clinical cases were identified as one of only four species: A. caviae (37.26%), A. dhakensis (23.49%), A. veronii (21.54%), and A. hydrophila (13.07%) (Fernández-Bravo and Figueras, 2020).
Diarrhea can be divided into inflammatory or noninflammatory, and both types have infectious and noninfectious causes. Infectious inflammatory diarrhea is often bacterial in etiology and may be related to travel or foodborne illness (Meisenheimer et al., 2022). Aeromonas spp. can cause short- or long-term diarrhea, but evidence for the enteropathogenic role of Aeromonas spp. had been controversial (Soltan Dallal et al., 2016). The pathogenicity was related to virulence factors, including aerolysin (Aer), heat-stable cytotoxin (Ast), hemolysin (HlyA), lateral flagella (Laf), polar flagellum (Fla), elastase (Ela), lipase (Lip), cytotoxic enterotoxin (Act), and cytotoxic enterotoxin (Alt) (Batra et al., 2016). Aeromonas spp. used these virulence factors to survive in the host, thus evading the host immune response and causing the host infection (Batra et al., 2016). Most Aeromonas spp. associated diarrhea patients were self-limiting; however, patients with serious diarrhea should receive an antimicrobial treatment. Up till now, most Aeromonas spp. have been observed as resistant to lincosamides, vancomycin, teicoplanin, and some penicillins, whereas tetracyclines, quinolones, chloramphenicol, and most of the aminoglycosides and the cephalosporins were initially reported to be effective against Aeromonas spp. (Kämpfer et al., 1999). However, extensive use of antibiotics in treatment increased resistance in Aeromonas spp. to antimicrobial drugs. Besides, there may be regional differences in drug susceptibility results (Liu et al., 2008). Therefore, it is necessary to perform drug susceptibility test in the local Aeromonas spp. isolates to establish an Aeromonas spp. associated drug resistance spectrum.
Vibrio parahaemolyticus, Salmonella spp., Shigella spp., and diarrheal Escherichia coli were routinely monitored in our region, but Aeromonas spp. weren’t; diarrhea caused by Aeromonas spp. was underappreciated in the clinic. Our study aims to show that Aeromonas spp. are present in our region and can cause diarrhea, especially during the warmer months. Therefore, increasing the level of hygiene in more areas and controlling bacterial foodborne diarrhea diseases should be given more attention by health officials.
Materials and Methods
Aeromonas isolates
All isolates (n = 41) in this study were collected from stool samples of diarrhea patients in the foodborne disease surveillance sentinel hospital of Wenzhou from 2019 to 2023. The automatic bacteriologic analyzer (VITEK 2 Compact, BioMèrieux, France) and the mass spectrometer (Matrix-assisted Laser Desorption Ionization Time-Of-Flight, Bruker, Germany) were used to identify the Aeromonas isolates, which were cultured at 36°C on RS agar plates (Hopebio, Qingdao, China).
Whole genome sequencing
DNA of Aeromonas isolates was extracted using a bacterial whole genome extraction kit (Tiangen, Beijing, China), and its concentration and purity were determined by spectrophotometer (NanoDrop One, Thermo Fisher, America). The sequencing strategy adopted second-generation sequencing, which interrupted DNA by enzyme and added specific joints to construct libraries. Libraries were determined by fluorimeter (Qubit, Thermo Fisher, America) for quality control and then paired-end sequenced by sequencer (Illumina MiSeq, America). The raw sequencing data were filtered, and the low-quality data were removed using Fastp tool. The clean sequencing data were spliced using SPAdes tool.
Species identification of Aeromonas isolates and multilocus sequence typing
The splicing data were uploaded to the official PubMLST website (http://pubmlst.org/) to obtain Aeromonas isolates species identification and their multilocus sequence typing (MLST), which relies on the sequencing of six housekeeping genes (gyrB, groL, gltA, metG, ppsA, and recA). A phylogenetic tree was also constructed using BioNumerics 7.6 (Applied Maths, Belgium).
Prediction of virulence gene and drug resistance gene
The splicing data were processed by abricate tool with the database from the official VFDB website (http://www.mgc.ac.cn/VFs/) and the official CARD website (https://card.mcmaster.ca/) to get virulence gene and drug resistance gene of Aeromonas isolates.
Antimicrobial susceptibility testing
Miniaturization of the broth dilution susceptibility test for Aeromonas spp. was performed following the Clinical and Laboratory Standards Institute guideline M45. And the CHNENF drug-sensitive plate (Thermo Fisher, America) was chosen to carry out drug susceptibility test. The minimal inhibition concentrations of nine antibiotics (cefotaxime [CTX], cefoxitin [CFX], ceftazidime [CAZ], imipenem [IPM], gentamicin [GEN], tetracycline [TET], ciprofloxacin [CIP], trimethoprim–sulfamethoxazole [SXT), and chloramphenicol [CHL]) were measured. E. coli ATCC 25922 was used for the quality control strains.
Statistics
Data analysis and Graph drawing were performed by GraphPad Prism 8 (GraphPad Software, America). Chi-square test and Fisher’s exact tests were performed to analyze for significant associations among seasons, as well as differences between subcategories of virulence genes and Aeromonas species. p-Values ≤0.05 were considered as significant.
Ethics statement
The studies involving human participants were reviewed and approved by the Wenzhou Center for Disease Control and Prevention. The patients/participants provided their written informed consent to participate in this study.
Results
The detection of Aeromonas isolates
From 2019 to 2023, a total of 1829 stool samples of diarrhea patients were collected from the sentinel hospital for detection (Supplementary Table S1), and the detection rate of Aeromonas spp. was 2.24% (41/1829). What’s more, the infection of Aeromonas spp. in foodborne diarrhea was always present, and the infection rate increased at first and then decreased from 2019 to 2023, with a peak detection rate (3.24%, 14/432) in 2021. In addition, our study showed that Aeromonas spp. is more easily detected in warmer months, especially in summer (from June to August, p < 0.05 [chi-square test]) (Fig. 1A). We identified all the Aeromonas isolates based on WGS, which were identified as follows: A. veronii (53.66%, 22/41), A. caviae (21.95%, 9/41), A. hydrophila (9.76%, 4/41), A. dhakensis (4.88%, 2/41), A. rivipollensis (4.88%, 2/41), A. enteropelogenes (2.44%, 1/41), and A. media (2.44%, 1/41) (Fig. 1B).

The distribution of Aeromonas isolates classified by
MLST of Aeromonas isolates
According to in silico sequence typing, all 41 strains can be divided into 38 sequence types (STs), 31 of which were novel (ST2487-ST2510 and ST2512-ST2517) (Fig. 2). Thus, Aeromonas isolates showed a high degree of genetic diversity. However, three isolates of A. hydrophila belonged to ST 574 (75%, 3/4), and all A. rivipollensis isolates belonged to ST 2510 (100%,2/2), revealing a high degree of homology.

Sequence types of Aeromonas isolates. A phylogenetic tree was constructed using the alleles of gyrB, groL, gltA, metG, ppsA, and recA to reveal the relationships between 41 Aeromonas isolates from foodborne diarrhea patients.
The distribution of virulence genes
A total of 259 virulence genes were found in the Aeromonas isolates, which can be divided into 5 major categories and 19 subcategories (Supplementary Fig. S1). These major categories were Adherence, Effector delivery system, Exotoxin, Motility, and Nutritional/Metabolic factor.
The category distribution of virulence genes in the Aeromonas strains is summarized in detail (Table 1). All species of Aeromonas isolates had MSHA type IV pili, Tap type IV pili, Exe T2SS, hemolysin HlyA, hemolysin III, thermostable hemolysin, polar flagella, and heme uptake system associated virulence genes. The distribution of Type I pili, T6SS, T6SS secreted effectors, aerolysin, extracellular hemolysin AHH1, heat-stable cytotonic enterotoxin Ast, RtxA, and amonabactin associated virulence genes was significantly different among the seven species of Aeromonas isolates [p < 0.05 (Fisher’s exact test)]. In addition, A. hydrophila had the largest subcategories of virulence genes (84.2%, 16/19).
The Categories Distribution of the Virulence Genes
Genotypic and phenotypic antimicrobial resistance
Fifty-seven antimicrobial resistance genes were searched in the Aeromonas isolates, 39 of which were associated with one class of antimicrobial agents (including aminoglycoside, carbapenem, cephalosporin, diaminopyrimidine, fluoroquinolone, fosfomycin, macrolide, peptide, phenicol, rifamycin, sulfonamide, and tetracycline). However, we also found multiple antimicrobial drug-related resistance genes, such as OXA, TRU-1, AAC(6′)-Ib-cr, VEB-1, MOX, TEM-1, VIM-1, PER-3, msrE, and MexD, which were associated with two or three or four or five or seven or nine classes of antimicrobial agents (Fig. 3). The aminoglycoside resistance gene accounted for 26.8% of Aeromonas isolates. The carbapenem resistance gene accounted for 70.7% of Aeromonas isolates. Moreover, the cephalosporin resistance gene accounted for 100% of Aeromonas isolates. In addition, the sulfonamide and diaminopyrimidine resistance gene (26.8%), the fluoroquinolone resistance gene (19.5%), the phenicol resistance gene (22.0%), and the tetracycline resistance gene (51.2%) were present in Aeromonas isolates.
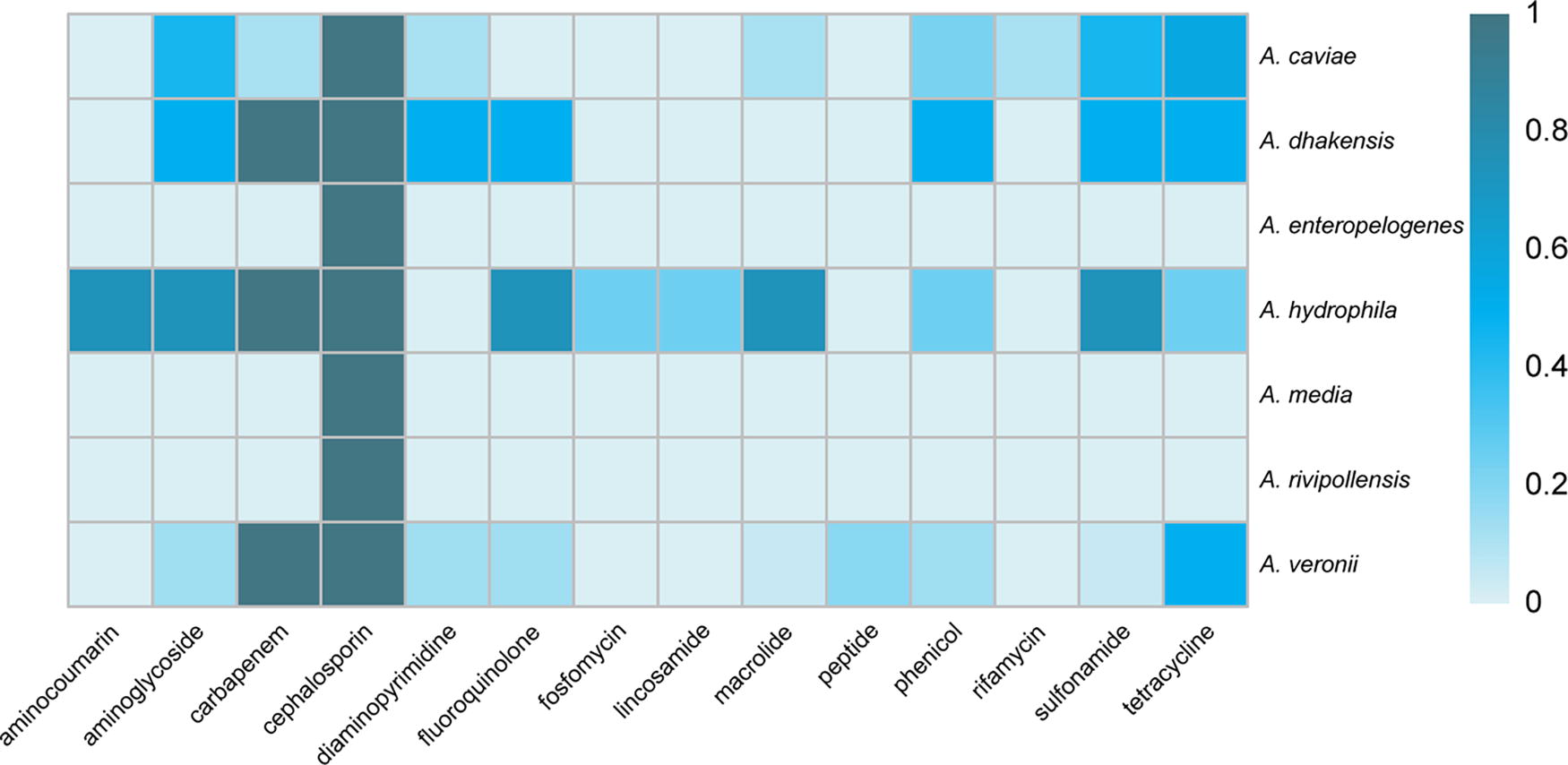
Distribution of putative antimicrobial resistance genes harbored by Aeromonas isolates in Wenzhou, 2019–2023.
A total of 37 Aeromonas isolates were tested for drug susceptibility. The antibiotic resistance rate in Aeromonas isolates to CTX, CFX, CAZ, IPM, GEN, TET, CIP, SXT, and CHL was 8.1%, 16.2%, 10.8%, 2.7%, 5.4%, 27%, 8.1%, 16.2%, and 2.7%. These 11 kinds of drugs belonged to 7 antimicrobial classes, for which in silico antimicrobial resistance gene detection was highly correlated with phenotypic antimicrobial resistance patterns with an overall sensitivity of 93.3% and a specificity of 66.7% (Table 2). With the exception of tetracycline, genotypic prediction of phenotypic resistance had a sensitivity of 100% for other classes. Meanwhile, aminoglycoside, tetracycline, fluoroquinolone, sulfonamide/diaminopyrimidine, and phenicol resulted in specificity >80%, whereas cephalosporin and carbapenem presented poor coherence between phenotypic resistance and the presence of corresponding resistance genes.
Discussion
Despite significant advances in health status and public health awareness, diarrhea remains one of the leading causes of disease burden worldwide. About 1.5 million deaths (ranking eighth as a common cause of life loss) due to diarrhea occurred worldwide in 2019 (WHO, 2020), having a huge impact on society. V. parahaemolyticus, Salmonella spp., Campylobacter spp., and Aeromonas spp. were the main bacterial pathogens causing foodborne diarrhea in Wenzhou. And Campylobacter spp. had the highest detection rate, whereas Aeromonas spp. had the second highest detection rate (Zhang et al., 2023). The detection rate of diarrhea-associated Aeromonas spp. was 2−10% in developed countries (Janda and Abbott, 2010) and 7% in Hong Kong, China (Chan et al., 2003). Besides, the detection rate of diarrhea-associated Aeromonas spp. in Wenzhou was 2.24%, which was similar in other regions. But we think that the actual detection rate of Aeromonas spp. should be higher. One of the best explanations is to try to understand that the reason for the current detection rate of Aeromonas spp. was the difficulty of isolation in Aeromonas spp. relative to Campylobacter spp. Thus, improving the isolation method of Aeromonas spp., such as cold enrichment, will be the direction of our future research.
Aeromonas spp. diarrhea cases have been reported worldwide. Verma et al. showed that pathogenic diarrheal agents were found in 63% of patients, and Aeromonas sp. was identified in 1% in North India (Verma et al., 2019). It was also reported that the detection rate of A. hydrophila was 1% from 200 collected samples in Arak, Iran (Abbasi et al., 2016). In addition, Shah et al. found that Aeromonas sp. was the second most frequently isolated pathogen (5.5%) of the 1410 hospitalized diarrheic children (Shah et al., 2016). All these studies suggested that diarrhea caused by Aeromonas spp. was a global problem and should be noted.
Among the 38 STs identified from the 41 Aeromonas isolates, 31 were novel STs, which indicated that the isolates obtained from stool samples of diarrhea patients had high genetic diversity. Qu et al. found 73 STs in 79 isolates obtained from food products, 65 of which were novel STs (Qu et al., 2022), whereas Lau et al. also found 36 STs in 47 isolates obtained from food products, 34 of which were novel STs (Lau et al., 2020). The results showed that Aeromonas spp. often had high genetic diversity, multiple clones, and various sources both in diarrhea patients and in food products. It is necessary to monitor the infection caused by these strains and conduct homology analysis in time to determine the source of infection and the route of transmission.
The pathogenicity of Aeromonas spp. was related to its virulence factors, and the pathogenic mechanism is complicated due to the synergistic effect of multiple virulence factors (Yu et al., 2005). Osman et al. also showed that the virulence factors such as Ast, HlyA, Alt, and AerA were significantly correlated with the virulence of A. hydrophila (Osman et al., 2012). Thus, detection of the virulence genes of Aeromonas spp. is very important for the prevention and control of diarrhea caused by Aeromonas spp. In our study, 259 virulence genes were found in the Aeromonas isolates. Among them, the common virulence gene HlyA was predicted in all Aeromonas isolates, Ast was detected in A. hydrophila and A. enteropelogenes strains, whereas AerA was only predicted in A. enterica strains, and Alt was not detected in any of the isolates. Besides, the distribution of several virulence gene subcategories was significantly different in the different species of Aeromonas isolates, such as Type I pili, T6SS, T6SS secreted effectors, aerolysin, extracellular hemolysin AHH1, heat-stable cytotonic enterotoxin Ast, RtxA, and amonabactin. These suggested that the distribution of virulence genes is species dependent.
A large number of studies have shown that Aeromonas spp. have a high resistance rate to ampicillin (Ko et al., 1996). In addition, ampicillin was added to the enrichment solution used to isolate Aeromonas strains in our study, so our study did not conduct drug susceptibility test to penicillins. Aeromonas spp. had different degrees of resistance to antibiotics, and tetracycline was the most serious, with a resistance rate of 27%. Therefore, the use of tetracycline should be avoided, and the appropriate antibiotics should be selected for the treatment of Aeromonas spp. infections according to the result of antimicrobial susceptibility test. In addition, we found that resistance genes detected in silico showed high-level concordance to phenotypic profiles in our previous study of Salmonella resistance (Fang et al., 2022). In addition, our study found that resistance genes of aminoglycoside, tetracycline, fluoroquinolone, sulfonamide/diaminopyrimidine, and phenicol detected in silico showed high-level concordance to phenotypic profiles in Aeromonas spp. resistance analysis. Thus, these results support the hypothesis that WGS is a powerful tool for predicting antimicrobial resistance genes in Aeromonas spp.
Conclusion
Aeromonas spp. was an important zoonotic pathogen, which can be found in the natural environment, animal, and human. Our study has shown that Aeromonas spp. in Wenzhou existed in seven species, and A. veronii was the most common species. Aeromonas spp. had high genetic diversity, and the 41 isolates in the current study were shown to produce 38 independent STs. Among virulence genes, HlyA gene was predicted in all Aeromonas isolates, Ast gene was detected in A. hydrophila and A. enteropelogenes strains, whereas AerA gene was only predicted in A. enteropelogenes strains, and Alt gene was not detected in any of the isolates. Therefore, the distribution of virulence genes is species dependent in our study. In addition, Aeromonas spp. had different degrees of resistance to antibiotics, and tetracycline was the most serious. In addition, we found that resistance genes of aminoglycoside, tetracycline, fluoroquinolone, sulfonamide/diaminopyrimidine, and phenicol detected in silico showed high-level concordance to phenotypic profiles in Aeromonas spp. resistance analysis. Our research highlighted the necessity for the development of prevention and control strategies to reduce the risk of foodborne diarrhea caused by Aeromonas spp.
Footnotes
Acknowledgment
The authors are grateful to Shengkai Li from Suzhou University for his technical assistance.
Authors’ Contributions
H.L.: Funding acquisition (lead), methodology (equal), project administration (lead), software (lead), visualization (equal), writing—original draft (equal), and writing—review and editing (lead). Y.L.: Conceptualization (lead), investigation(lead), visualization(equal), software (supporting), and writing—review and editing (supporting). Y.W.: Data curation (lead), formal analysis (lead), resources (supporting), and writing—original draft (equal). Y.H.: Methodology (equal) and writing—original draft (equal). L.Z.: Conceptualization (supporting), methodology (equal), supervision (lead), validation (lead), and writing—review and editing (supporting).
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by the Science and Technology Project of Wenzhou (Y20220298).
Supplementary Material
Supplementary Figure S1
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.