
Abstract
Staphylococcus aureus (S. aureus) is a pathogen capable of causing severe diseases and exhibiting resistance to multiple antibiotics. However, there is a significant lack of comprehensive research on the global prevalence of its antibiotic resistance genes (ARGs). This study provided a comprehensive analysis of ARGs in S. aureus, using 113,842 S. aureus genome sequences from the National Center for Biotechnology Information database. The results revealed that a significant majority (84%) of these genomes harbored at least one ARG, with a total of 389,464 ARG sequences identified across 19 major types and 103 subtypes. These ARGs exhibited varied abundances and diversities, linked primarily to clinical cases worldwide. ARGs for fluoroquinolones, multidrug resistance, bacitracin, tetracyclines, beta-lactams, and aminoglycosides were notably abundant, ranging from 3.16 × 10 − 5 to 1.49 copies of ARG per million bp. Variations in the abundance and diversity of ARGs were observed between countries, with middle- and low-income countries showing higher gene abundance but lower diversity compared with high-income countries. Temporal analysis over 30 years showed a fluctuating decline in ARG abundance alongside an increase in diversity, suggesting evolving resistance mechanisms. The study also explored the role of mobile genetic elements in ARG dissemination, finding a substantial proportion of ARG subtypes associated with plasmids and insertion sequence elements, indicating their potential for spread across borders. The global distribution of mobile ARGs was further analyzed, revealing the extensive reach of certain ARGs across countries. This research provides valuable insights into the prevalence and dissemination of antibiotic resistance in S. aureus on a global scale, aiding in the development of effective monitoring and control strategies to combat ARGs in S. aureus and other pathogens.
Introduction
The widespread use and misuse of antibiotics in humans and animals have generated significant selective pressure, accelerating the rapid development of microbial resistance to drugs. Antibiotic resistance in bacteria has evolved into a global public health emergency. The Centers for Disease Control and Prevention (CDC) highlighted in its 2019 Antibiotic Resistance Threats Report that ∼2.8 million Americans contract antibiotic-resistant bacterial infections annually, leading to around 35,000 deaths (CDC, 2019). The World Health Organization (WHO) predicts that, without effective measures, the global annual death toll from antibiotic-resistant bacterial infections could reach 10 million by 2050 (O’Neill, 2016). However, research on resistance mainly focuses on short-term clinical settings (Gan et al., 2021; Xu et al., 2020). Studies on the long-term trends of antibiotic resistance are relatively scarce.
Staphylococcus aureus (S. aureus) is widely distributed in the natural environment and the normal skin microbiota of animals and humans (Heaton et al., 2020; Zhou et al., 2023a). Studies have shown that about 20–30% of the population are long-term carriers of S. aureus, and although most do not exhibit clinical symptoms, this bacterium remains a significant human pathogen (Hanselman et al., 2009; Humphreys, 2012; Wertheim et al., 2005; Zhou et al., 2023b). S. aureus-related bacterial infections caused 1.1 million deaths in 2019, making it the most severe form of bacterial infection globally after ischemic heart disease (Ikuta et al., 2022). Research also indicates that about 15–20% of the S. aureus genome consists of mobile genetic elements (MGEs), which are closely associated with the spread of resistance in S. aureus (Lindsay, 2014). As S. aureus antibiotic resistance increases, clinical treatment faces greater challenges, especially with the emergence of methicillin-resistant S. aureus (MRSA), making the treatment of infections caused by this pathogen more difficult. The WHO has classified it as a high-priority pathogen for the development of new antibiotics (Pennone et al., 2022). Despite this, current research on S. aureus resistance is mainly focused on clinical settings or limited to specific global regions, with insufficient studies on the differences in resistance levels of S. aureus strains across different countries, the global spread of antibiotic resistance genes (ARGs), and the long-term resistance trends of S. aureus.
Given the significant gap in comprehensive understanding and long-term data on S. aureus resistance, this study is founded on the hypothesis that global genomic analysis can illuminate the dynamics of antibiotic resistance, particularly the roles and prevalence of ARGs and MGEs in S. aureus from different geographical locations over time. By testing this hypothesis, we aim to provide crucial insights into the trends of antibiotic resistance in S. aureus on a global scale, thus informing the development of more effective monitoring and control strategies for antibiotic resistance. This approach is not only critical for understanding the current landscape of microbial resistance but also imperative for preempting future challenges in public health management.
Materials and Methods
Data collection
All genomic sequences of S. aureus were retrieved from the “Nucleotide” database on the National Center for Biotechnology Information (NCBI) official website (https://www.ncbi.nlm.nih.gov) using the keyword “Staphylococcus aureus” (as of April 16, 2023). The criteria for filtering sequences for subsequent analysis are detailed in the Supplementary Data S1. Institutional Review Board (IRB) approval was not required because this article is a meta-analysis. The data comes from public database and does not require ethical approval.
Identification of ARGs
The Prodigal (v2.6.3) software was used for functional gene prediction on the downloaded S. aureus genome sequences, retaining the predicted genes and amino acid sequences. The predicted amino acid sequences were compared with the Integrated Antibiotic Resistance Database (Liu et al., 2019), which incorporates known ARGs from databases such as Comprehensive Antibiotic Resistance Database (CARD), Antibiotic Resistance Genes Database (ARDB), and NCBI, removing duplicates and erroneous sequences and categorizing ARGs. Sequences with a similarity of ≥80% and coverage of ≥70% were identified as ARGs (Hu et al., 2013).
Diversity and abundance of ARGs
The diversity of ARGs is quantified by the number of ARG subtypes present, reflecting the variety of ARG subtypes within S. aureus genomes sourced from various countries and regions. The abundance of each resistance gene was represented as “copies of ARG per million bp” (CPM), calculated using the formula:
Here,
Identification of MGE-associated ARGs
S. aureus genome sequences carrying ARGs were extracted using a self-written Perl script. Plasmid genome sequences were identified from the GenBank files. In addition, the sequences were compared with the insertion sequence (IS) transposase database using BLAST software. The references in IS transposase database were collected from the IS Database (ISfinder) (Varani et al., 2011) and the NCBI nr database. Sequences with a similarity of ≥80% and coverage of ≥70% were identified as IS. ARGs located within three genes or 5000 bp of IS sequences were classified as IS-associated ARGs (Ellabaan et al., 2021). Both plasmid-located and IS-associated ARGs were categorized as MGE-related ARGs.
Global distribution of MGE-associated ARGs
A self-written Perl script was used to extract ARG sequences associated with MGEs. USEARCH (v10.0.240) software was then used to eliminate duplicate sequences, forming an S. aureus mobile ARG database. Using BLAST software and combining sequence GenBank file information, ARG sequences from different countries were compared with the above database. A 99% gene homology threshold was used to reveal the global distribution and spread of identical or highly homologous mobile ARG sequences.
Results
Occurrence, abundance, and diversity of ARGs in S. aureus
As of April 16, 2023, an analysis of 113,842 S. aureus genome sequences collected from the NCBI database revealed that 84% harbored at least one ARG. A total of 389,464 ARGs were identified from S. aureus genomes collected globally from 80 countries between 1900 and 2022, most of which were directly associated with human clinical cases (Supplementary Table S1). These encompassed 19 major types and 103 subtypes of ARGs. There were significant differences in the abundance of resistance types, with fluoroquinolones, multidrug resistance (MDR), bacitracin, tetracyclines, beta-lactams, and aminoglycosides showing relatively higher abundances, constituting 83.28% of the total ARG abundance within the S. aureus genomes. Among the subtypes, norB, mdeA, mepA, mgrA, norA, bceA, bacA, tetL, mepR, FmtA, blaDHA-1, apH-Stph, aacA, and dfrG were detected with higher abundance, ranging from 0.38 to 1.49 CPM. The norB gene was the most prevalent, accounting for 12.19% of all identified ARGs, followed by becA and tetL genes (Fig. 1).
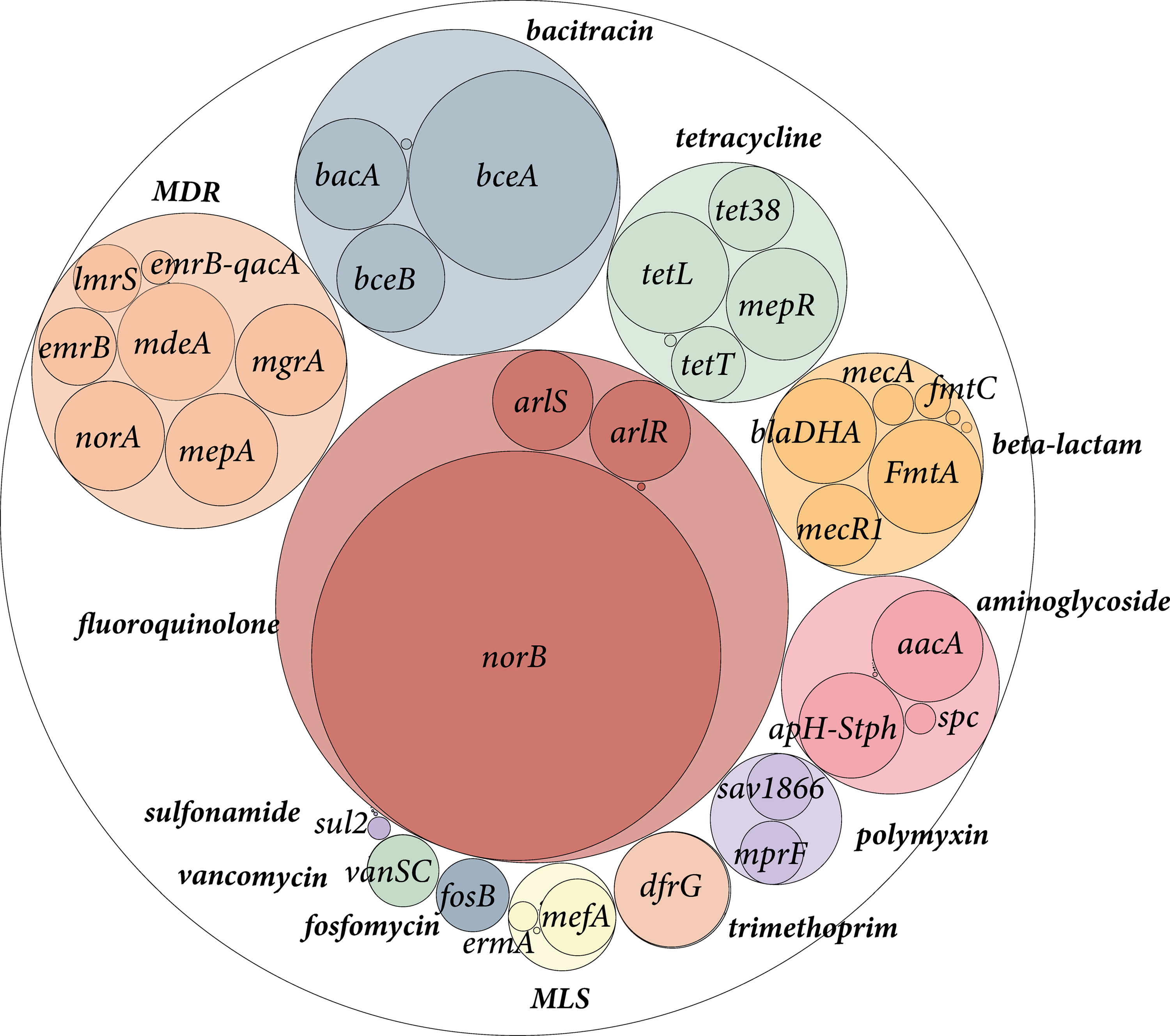
Global distribution and abundance of ARGs in Staphylococcus aureus. Colored by ARG type. Outer and inner circles represent ARG types and subtypes, respectively. Circle size is proportional to the abundance of ARGs. ARGs, antibiotic resistance genes; MDR, multidrug resistance; MLS, macrolide–lincosamide–streptogramin.
Distribution of ARGs in different countries
Analysis of ARG abundance and diversity in the top 15 countries with the number of S. aureus genome sequences revealed differences between countries. Malaysia exhibited the highest gene abundance at 15.16 CPM, followed by Ghana, China, Italy, Tanzania, and India, whereas Spain and France had relatively lower abundances (Fig. 2a). The United States had the highest diversity of ARGs, reaching 81 subtypes, whereas Thailand and Ghana showed lower diversity (Fig. 2c). Rarefaction curves indicate that the current sample sizes were adequate for analysis (Fig. S1a).

Comparative analysis of the abundance
A total of 96 ARG subtypes across 18 resistance categories were identified in 15 countries, with significant variation in gene abundance among different countries, ranging from 0 to 1.87 CPM (Supplementary Table S2). Sixty ARGs had an abundance greater than 1 × 10 − 3 CPM in at least one country sample. ARGs associated with bacitracin, beta-lactams, fluoroquinolones, MDR, and tetracyclines had relatively high abundances across the 15 country samples (Supplementary Data S1). Thirty-two ARG subtypes were common across all country samples. Specifically, norB and tetL dominated in these countries, with average abundances of 1.67 ± 0.12 CPM and 0.51 ± 0.06 CPM, respectively (Supplementary Table S3; Supplementary Data S1).
Temporal dynamics of ARGs
To explore the temporal changes in ARGs carried by S. aureus, 78,761 genome sequences isolated between 1993 and 2022 were analyzed. Rarefaction curves indicated that the sample sizes for each time period were adequate (Fig. S1b). Notably, over the past 30 years, the global abundance of ARGs in S. aureus showed a fluctuating decline, whereas diversity gradually increased until a drop in 2018–2022 (Fig. 3a). Moreover, the diversity of identified ARG subtypes increased from 43 in 1993–1997 to 71 in 2013–2017 and 67 in 2018–2022 (Fig. 3b). Over these 30 years, S. aureus carried ARGs primarily belonging to aminoglycosides, bacitracin, beta-lactams, fluoroquinolones, MDR, and tetracyclines. Compared with other ARG subtypes, aacA, bceA, FmtA, and norB genes maintained high and relatively stable abundances across the six time spans (Supplementary Table S4; Supplementary Data S1).
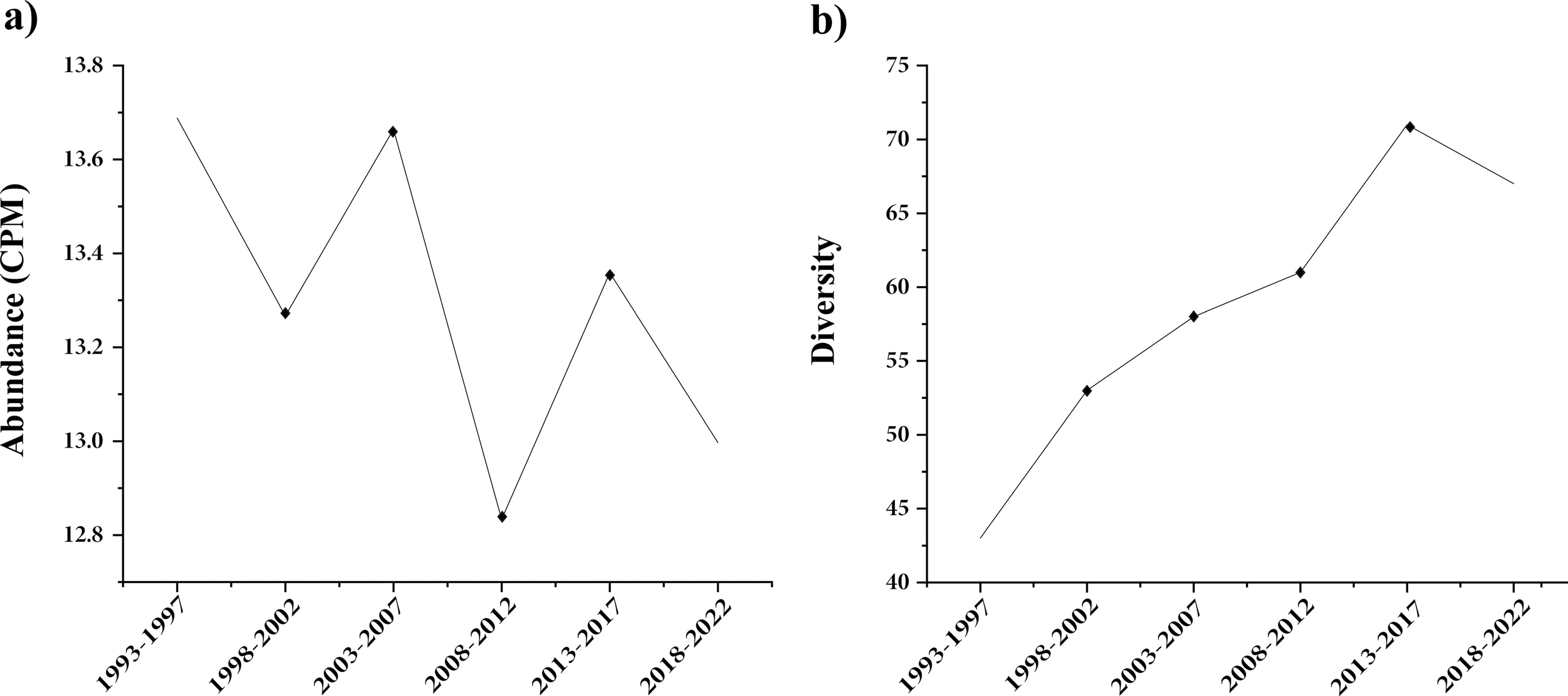
Temporal analysis of the abundance
MGE-related ARGs
A total of 148,006 IS elements and 1475 plasmid sequences were identified within the S. aureus genomes, with IS1182 being the most significant, accounting for 51% of all IS elements. Further analysis revealed that ∼3.95% of identified ARG sequences were associated with these MGEs, covering 19 different resistance categories. Among these MGE-related ARGs, 11,187 were associated with IS elements, and 4227 were plasmid associated. Furthermore, these MGE-related ARGs accounted for 3.87% of the abundance among all ARGs. Of the 103 identified ARG subtypes in S. aureus, 87.38% were associated with MGEs, with plasmid-associated genes comprising 72.82%, IS-associated genes 56.31%, and genes associated with both types of elements 42.72%, whereas only 12.62% of ARG subtypes were not associated with MGEs (Supplementary Table S5; Fig. 4).

Venn diagram showing the shared and unique ARG subtypes across different genetic backgrounds. ARG, antibiotic resistance gene.
Global distribution of mobile ARGs
Analysis of the distribution of mobile ARGs across different countries, using 99% homology clustering, yielded 733 operational taxonomic units (OTUs) of ARGs. Of these, 11% were present in five or more countries (Supplementary Table S6). The United States, China, Germany, and Australia shared a higher number of MGE-related ARGs. Fluoroquinolone, beta-lactam, and trimethoprim genes were relatively widely distributed. Two mecR1 genes and one blaPC gene, three norB genes, and one dfrG gene had the widest distribution and were present in over 39% of the countries (Fig. 5).
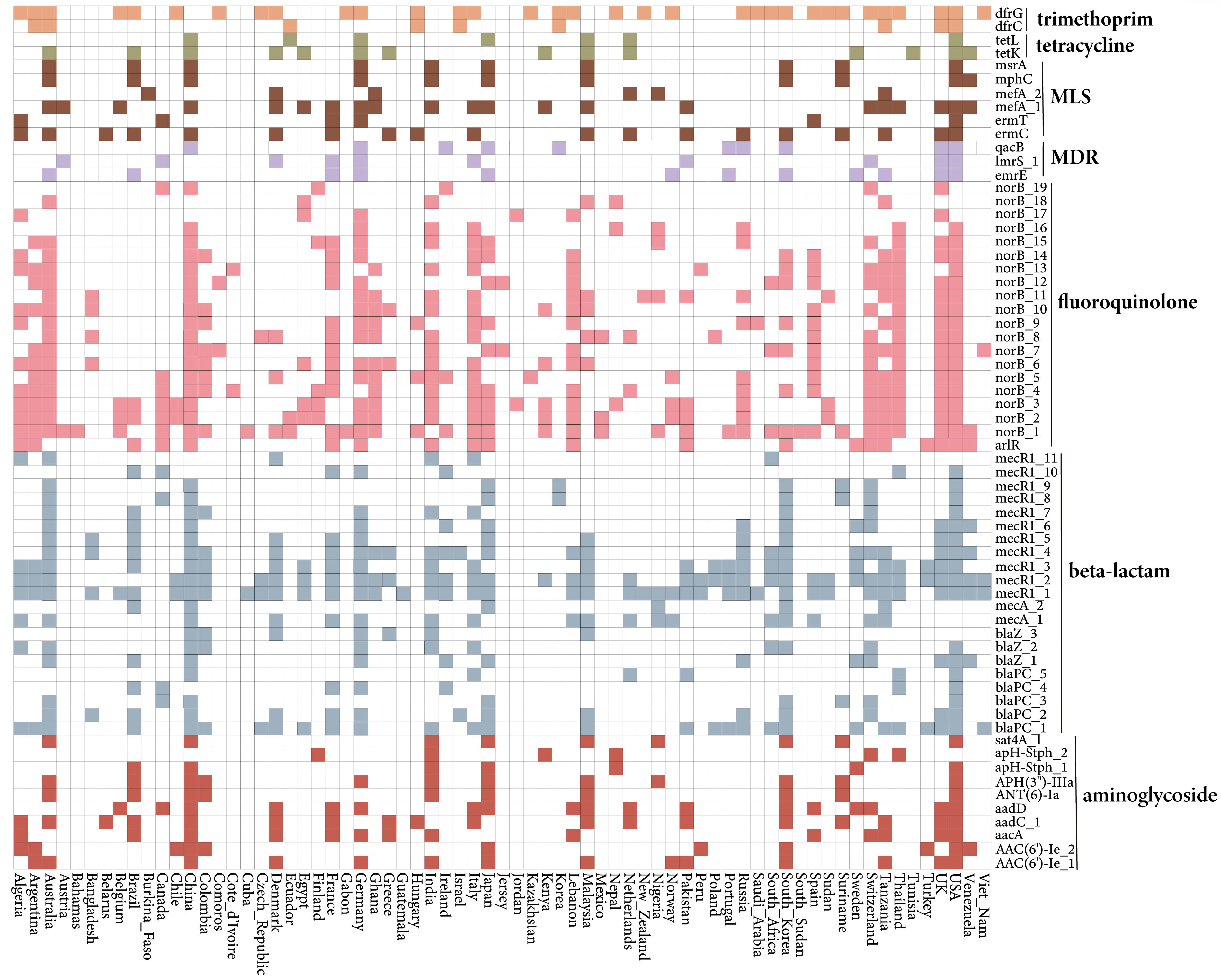
Global distribution of major mobile ARGs in Staphylococcus aureus. Only the mobile ARGs present in six or more countries are shown. The presence of ARGs in each country is denoted by colored squares, whereas their absence is indicated by white squares. ARGs, antibiotic resistance genes.
To visually represent the global spread of mobile ARGs, the three highest abundance ARG OTUs—norB, mecR1, and dfrG—were analyzed for global distribution. These three ARGs were widely distributed, with 77% of countries having at least one of these genes and 28% of countries harboring all three (Supplementary Table S6). The United States, China, the United Kingdom, Italy, and Switzerland had relatively high abundances of these three ARGs. Overall, these three ARGs were primarily distributed in Europe, Asia, and Africa. norB and mecR1 genes were distributed across all continents except Antarctica, with norB mainly in Europe, Asia, and Africa, and mecR1 primarily in Europe, Asia, and South America. In contrast, dfrG was distributed in five continents, excluding Antarctica and North America, mainly in Asia, Europe, and Africa (Fig. 6).

Global distribution of the top three abundant mobile ARG OTUs in Staphylococcus aureus. Only countries and regions containing a number of ARG OTUs ≥5 are annotated on the map. ARG, antibiotic resistance gene; OTUs, operational taxonomic units.
Discussion
S. aureus demonstrates a remarkable ability to adapt to various environmental conditions and rapidly acquire resistance to these antibiotics (McCallum et al., 2010). Among the analyzed genome sequences, 84% contained ARGs, showcasing a high diversity with 19 major classes and 103 subtypes. This indicates S. aureus’s diverse adaptive responses to antibiotic pressure and underscores the need for cautious management of these antibiotics. ARGs associated with commonly used antibiotics, including aminoglycosides, beta-lactams, fluoroquinolones, and tetracyclines, were identified with high prevalence, similar to previous studies (Zaghen et al., 2023). The assessment of ARGs in S. aureus from human, environmental, food, and animal sources revealed significant differences in ARG diversity and abundance. Human-derived S. aureus exhibited the highest diversity, with 97 distinct ARG subtypes (Supplementary Table S7; Fig. S5a), likely because of the extensive use of antibiotics in human medicine. In contrast, animal-derived S. aureus showed a relatively high abundance of bacitracin, sulfonamide, and tetracycline resistance genes (Supplementary Table S8), which can be attributed to the widespread use of these antibiotics in livestock farming. At present, vancomycin is used as the first-line treatment for MRSA (Li et al., 2023). However, its related ARGs also contribute to the global ARG abundance of S. aureus (Fig. 1). Studies have shown that the global prevalence of vancomycin resistant S. aureus (VRSA) has been steadily increasing (Shariati et al., 2020), indicating that monitoring resistance and complying with infection control guidelines are necessary to prevent further spread and maintain effectiveness of vancomycin.
The consumption and misuse of antibiotics are key drivers of antimicrobial resistance (Barker et al., 2017; Malhotra-Kumar et al., 2007). This study found that the abundance of ARGs in S. aureus from middle- and low-income countries was significantly higher compared with high-income countries (Fig. 2b; analysis of variance, p = 0.0041), aligning with previous findings where resistance rates for common pathogens, including S. aureus, were higher in lower-income settings (Klein et al., 2019). This disparity may relate to the rapid increase in antibiotic consumption and the lack of effective antibiotic stewardship in these countries. For example, India showed a high abundance of ARGs, with a massive volume of antibiotic sales in the private sector, including drugs banned by the government, and relatively weak antibiotic regulation (Mehta et al., 2022). Poor environmental hygiene and sanitation facilities in resource-limited countries also contribute to the development of resistance (Cairncross, 2003). Despite differences in the abundance of ARGs across countries, there was consistency in the prevalence of certain resistant types, such as beta-lactams, polymyxins, fluoroquinolones, multidrug resistance, and tetracyclines, reflecting global usage patterns and similar selective pressures.
Conversely, this study revealed that high-income nations generally exhibit a higher diversity of ARGs than middle- and low-income countries (Fig. 2d). Such disparities in diversity may be linked to the health care policies and practices as well as surveillance mechanisms of different countries. The United States has gradually established a stringent regulatory framework against antibiotic-resistant bacteria and has developed a 5-year national action plan to accelerate the research and development of new antibiotics, vaccines, and other therapeutic methods (Bright-Ponte et al., 2019; Hayes, 2022). These countries can adopt rapid responses when high antibiotic resistance emerges, potentially leading to an increase in the diversity of ARGs while effectively controlling their abundance. Conversely, because of economic constraints, middle- and low-income countries may rely more on basic and commonly used antibiotics, with less use of more expensive or specialized second-line drugs (Mendelson et al., 2016), resulting in relatively lower diversity of ARGs for specific pathogens.
Over the past 30 years, the abundance of ARGs in S. aureus has overall shown a fluctuating downward trend. This may reflect the implementation of stricter global guidelines for antibiotic use and infection control measures and improved antibiotic resistance monitoring and management strategies worldwide. For example, countries like France and Switzerland have initiated national campaigns to reduce the use of nonessential or inappropriate antibiotics, effectively reducing antibiotic consumption (Sabuncu et al., 2009). India launched a national policy against microbial resistance in 2011 (Wattal and Goel, 2014). Notably, certain ARGs (e.g., aacA, bceA, bacA, FmtA, and norB) have shown high and relatively stable abundance over the analyzed period. This stability might reflect the importance of these ARGs for the survival and spread of antibiotic-resistant S. aureus and their continued use with specific antibiotic classes. The stable abundance may also indicate that, despite efforts to reduce antibiotic use, specific types of resistance are still being maintained to some extent.
Horizontal gene transfer plays a crucial role in bacteria acquiring new pathogenic and antibiotic resistance features, with MGEs, such as IS and plasmids, facilitating the rapid acquisition of these traits, supporting bacterial survival in changing environments (Fang et al., 2023). This study found that ARG-containing S. aureus carried a significant number of MGEs. Importantly, 66.24% of IS-related ARGs were associated with IS1182, suggesting a dominant role for IS1182 in the spread of resistance. IS elements promote resistance expression through gene inactivation or activation, such as the insertion of IS1182 leading to the inactivation of the lytH gene, increasing the methicillin resistance levels in MRSA clinical isolates, and promoting the frequent occurrence of high-level MRSA strains (Fujimura and Murakami, 2008). Moreover, MGE-associated ARGs exhibited high diversity, covering 90 subtypes of all major resistance classes identified in this study, indicating the strong potential of MGEs for spreading resistance to a wide range of antibiotics, posing challenges to the control and treatment of S. aureus infections.
Globalization and increased international travel have exacerbated the spread of multidrug-resistant bacterial strains worldwide. Wildlife (Wang et al., 2017), fish (Almeida et al., 2017), and legally or illegally imported meats (Campos et al., 2018; Jansen et al., 2019) can also serve as carriers of resistant bacteria, facilitating cross-border transmission. This study showed that S. aureus’s mobile ARGs have widely disseminated globally, suggesting that S. aureus carrying ARGs may be spread through routes such as international travelers and trade goods. In addition, the geographical distribution of S. aureus mobile ARGs varied among countries such as the United States, China, and the United Kingdom, in terms of not only rich diversity but also dense distribution, indicating a higher risk of spread. ARGs for fluoroquinolones, beta-lactams, and trimethoprim were more widely spread than other ARGs, with norB, mecR1, and dfrG showing the highest OTU abundance globally, indicating a very high potential for spread and the need for vigilance regarding their potential to diminish treatment efficacy. Particularly, nearly 98% of trimethoprim ARG abundance was contributed by the dfrG gene, whose plasmid-encoded nature facilitates the spread of resistance (Reeve et al., 2019), necessitating focused monitoring of these ARGs to limit their spread.
Conclusion
This study emphasizes the importance of monitoring and controlling antibiotic resistance in S. aureus and other pathogens, highlighting the connection between ARGs and MGEs and the risk of global dissemination. Further research is needed to understand resistance mechanisms in different contexts and design effective monitoring and control strategies. Enhanced surveillance and preventative measures should target high-risk ARGs, such as fluoroquinolone, beta-lactam, and trimethoprim ARGs, and international cooperation and data sharing are crucial to combat antibiotic resistance globally.
Footnotes
Acknowledgments
The authors express their sincere gratitude to Xiaofang Huang and Xiaoyan He for their technical assistance. Special appreciation is also extended to Quanzeng Li and Qi Liao for their invaluable critical review of the manuscript.
Authors’ Contributions
G.J.: Formal analysis (equal), visualization (lead), writing—original draft (lead), and writing—reviewing and editing (equal). K.L.: Formal analysis (equal) and resources (equal). Y.Q.: Formal analysis (equal). L.Q.: Formal analysis (equal). Z.Z.: Supervision (equal). Z.L.: Conceptualization (lead), writing—reviewing and editing (equal), and supervision (equal).
Author Disclosure Statement
The authors declare that they have no conflict of interest.
Funding Information
This work was supported by the Science and Technology Base and talent Special Project of Guangxi (Grant No. 2022AC21335), the Natural Science Foundation of Xiamen, China (3502Z20227231), and the National Natural Science Foundation of China (Grant No. 32360039).
Supplementary Material
Supplementary Data S1
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.