
Abstract
Variants of glutathione S-transferase M1 (GSTM1) and T1 (GSTT1) genes have been implicated as risk factors for chronic myeloid leukemia (CML). However, the genetic association studies that examined the relation between the null genotypes of GSTM1 and GSTT1 genes and risk of developing CML gave conflicting or inconclusive results. In an attempt to interpret these results, a meta-analysis of all available studies (nine studies, with 757 cases and 1959 controls) was performed. In the meta-analysis the pooled odds ratios (OR) were estimated using random effects models. The heterogeneity between studies, the sources of potential bias, and the consistency of genetic effects across ethnicities were explored. Cumulative meta-analysis was also performed. Overall, the meta-analysis showed nonsignificant association between GSTM1 null genotype and CML (OR = 1.00 [0.83-1.20]) and lack of heterogeneity between the studies (pQ = 0.87). The association was also nonsignificant in Whites, East Asians, and Indians: OR = 1.38 (0.43-4.46), 0.94 (0.65-1.35), and 1.16 (0.74-1.82), respectively. However, GSTT1 null genotype was associated with increased risk of CML (OR = 1.57 [1.13-2.17]) and the heterogeneity between studies was significant (pQ = 0.04). In Indians, the association was significant (OR = 2.89 [1.56-5.35]) whereas in East Asians it was not significant (OR = 1.07 [0.74-1.54]). The combined GSTM1 normal/GSTT1 null genotypes produced significant association (OR = 1.95 [1.17-3.24]). Cumulative meta-analysis for GSTT1 gene showed an upward trend in risk effect, whereas the trend was downward in GSTM1. There was a differential magnitude of effect in large versus small studies. In conclusion, the accumulated evidence indicated an association between GSTT1 null genotype and CML.
Introduction
C
Glutathione S-transferases (GSTs) are a group of enzymes with complex multigenetic nature involved in phase II metabolism, catalyzing the conjugation of glutathione, and detoxification of carcinogens (Mondal et al., 2005). GSTM1 and GSTT1 are polymorphic genes that encode GSTs. In both genes there are polymorphisms with deletion of the entire gene. Individuals with homozygous deletions (null genotype) in both genes are supposed to have less ability to metabolize carcinogens and may therefore be more susceptible to cancers (Chen et al., 2008). Heterozygotes for the deletion are presumably normal and produce sufficient amounts of enzyme (Mondal et al., 2005). However, it is possible for individuals to possess a deletion on one chromosome and another type of mutation on the other. Thus, GSTM1 and GSTT1 may be considered candidate genes for CML susceptibility.
The genetic studies that have examined whether variants in the GSTM1 and GSTT1 genes are associated with CML have yielded conflicting or inconclusive results, possibly due to small sample sizes or the use of different populations. In an attempt to interpret these contradictory results and to overcome the aforementioned limitations, a comprehensive meta-analysis of all available studies relating the variants of GSTM1 and GSTT1 genes to the risk of CML was carried out (Zintzaras and Lau, 2008a, 2008b). In addition, heterogeneity between studies and potential bias were explored (Zintzaras and Ioannidis, 2005; Zintzaras and Lau, 2008a). Cumulative and recursive cumulative meta-analyses were also performed in order to investigate the trend of association as evidence accumulated (Lau et al., 1992; Zintzaras and Lau, 2008a).
Methods
Selection of studies
All studies published before 1 January 2009 were identified through extended computer-based search of PubMed database. As a search criterion, combinations of the following keywords were used: “glutathione S-transferase,” “GSTM1,” “GSTT1,” “chronic myeloid leukemia,” “chronic myeloid,” “chronic myelocytic leukemia,” “chronic myelogenous leukemia,” “chronic granulocytic leukemia,” “myeloid leukemia,” “myelogenous leukemia,” “CML,” “gene,” “polymorphism,” “association,” “risk,” and “susceptibility.”
All references cited in the studies were also reviewed to identify additional published work that was not indexed by the search in PubMed. Case reports, editorials, and review articles were excluded. Non-English articles were also excluded.
Case-control studies that determined the frequency of null and normal genotypes of GSTM1 or GSTT1 genes in patients with CML (cases) and controls were eligible for inclusion in the meta-analysis. Only studies carried out in human subjects using standard genotyping methods were considered. Family studies were excluded owing to different design considerations.
Data extraction
From each study the following information was extracted: first author, journal, year of publication, ethnicity of study population, age, gender, matching, genotyping method, and the number of cases and controls for each genotype (null and normal). Whether the genotypic data had been read with blinding disease status was also recorded. Where studies involved the investigation of more than one gene, information on combined genotypes was also recorded.
Data synthesis
The meta-analysis examined the association between null genotype and risk of CML development compared with normal genotype for GSTM1 and GSTT1 genes. The combined effect of the two genes (GSTM1 null/GSTT1 null vs. GSTM1 normal/GSTT1 normal) in CML was also examined.
In each individual study, the associations were indicated as odds ratios (OR) with the corresponding 95% confidence interval (CI). Subsequently, a pooled OR was estimated based on the individual ORs (Trikalinos et al., 2008; Zintzaras and Lau, 2008a). The heterogeneity between studies was tested using the Q-statistic (Zintzaras and Lau, 2008a). Heterogeneity was considered statistically significant if pQ < 0.10. Heterogeneity was quantified with the I2 metric, which is independent of the number of studies included in the meta-analysis (Zintzaras and Lau, 2008a). I2 measures values between 0% and 100% with higher values denoting a greater degree of heterogeneity. The pooled OR was estimated using random effects (RE) model (DerSimonian and Laird, 1986). RE modeling assumes a genuine diversity in the results of various studies incorporating study variance. When there is lack of heterogeneity the RE model coincides with the fixed effects model (Zintzaras and Lau, 2008a).
Cumulative and recursive cumulative meta-analyses were carried out for each gene to evaluate the trend of RE OR over time (Lau et al., 1992; Zintzaras and Lau, 2008a). In cumulative meta-analysis, studies were first chronologically ordered by publication year, and the pooled ORs were then obtained at the end of each year, that is, at each information step. This analysis shows the evolution of summary effect (OR) over time. In recursive cumulative meta-analysis, the relative change in pooled OR at each information step (OR in next year/OR in current year) was calculated (Zintzaras and Lau, 2008a). Thus, cumulative and recursive cumulative meta-analyses provide a framework for updating a genetic effect from all studies and a measure of to what extent this genetic effect alters as evidence accumulates (Zintzaras and Lau, 2008a).
The differential magnitude of effect in large versus small studies was assessed using the Egger regression test (Egger et al., 1997). Given that this test is underpowered, it was considered statistically significant for p < 0.10. In addition to the main (overall) analysis, which included all available data, a subgroup analysis for each ethnicity was performed (Zintzaras and Hadjigeorgiou, 2005; Zintzaras and Lau, 2008a). Analyses were performed using CUMAGAS (http://biomath.med.uth.gr) and Compaq Visual Fortran90 with International Mathematics and Statistics Library.
Results
Eligible studies
The literature review identified 45 articles in PubMed that met the search criteria. The full articles and their references were analyzed to assess their appropriateness for meta-analysis. Data from nine articles (studies) met the inclusion criteria and were included in the meta-analysis (Lemos et al., 1999; Löffler et al., 2001; Hishida et al., 2005; Lourenco et al., 2005; Mondal et al., 2005; Bajpai et al., 2007; Chen et al., 2008; Souza et al., 2008; Taspinar et al., 2008). The articles had been published between 1999 and 2008. Details abstracted from these studies included in the meta-analysis are provided in Table 1.
MTHFR, methylenetetrahydrofolate reductase; GSTM1, glutathione S-transferase M1; GSTT1, glutathione S-transferase T1; CML, chronic myeloid leukemia.
All the above nine studies dealt with the GSTM1 gene, and eight of these (except Lemos et al.) also dealt with the GSTT1 gene. Four studies provided data regarding combined genotypes of GSTM1 and GSTT1 genes (Lourenco et al., 2005; Bajpai et al., 2007; Souza et al., 2008; Taspinar et al., 2008). But one study provided data only for combined null versus normal genotypes (Lourenco et al., 2005). The studies were conducted in various populations with different ethnic groups: two involved East Asians (Hishida et al., 2005; Chen et al., 2008), two Whites (Lemos et al., 1999; Löffler et al., 2001), two Indians (Mondal et al., 2005; Bajpai et al., 2007), one Turkish (Taspinar et al., 2008), and two mixed population (Lourenco et al., 2005; Souza et al., 2008). The investigators in three studies stated that controls were ethnically and/or geographically matched (Lourenco et al., Mondal et al., Bajpai et al.). All studies reported that standard genotyping methods were used. None of the studies provided genotypes according to age or gender.
Summary statistics
The studies provided 757 cases and 1959 controls for GSTM1 and 746 cases and 1833 controls for GSTT1. The prevalences (%) of GSTM1 null and GSTT1 null genotypes in cases/controls were 44%/46% and 29%/28%, respectively. The prevalence of the combined GSTM1 null/GSTT1 null genotype was 19% in cases and 11% in controls.
Main results, subgroup and sensitivity analyses
The results for the association between the GSTM1 and GSTT1 genotypes and the risk of CML are shown in Table 2 and Figures 1 and 2.

Random effects (RE) odds ratio (OR) estimates with the corresponding 95% confidence interval (CI) for (
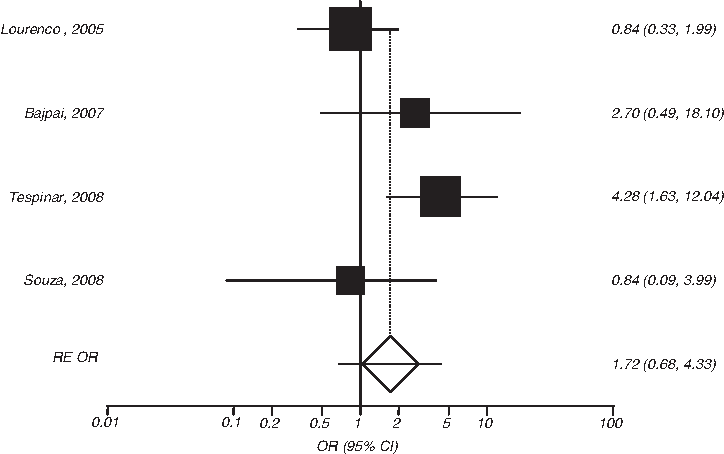
RE OR estimates with the corresponding 95% CI for the combined GSTM1 null/GSTT1 null vs. GSTM1 normal/GSTT1 normal genotype contrast and the risk of CML. The OR estimate of each study is marked with a solid black square. The size of the square represents the weight that the corresponding study exerts in the meta-analysis. The CIs of pooled estimates are displayed as a horizontal line through the diamond; this line might be contained within the diamond if the CI is narrow. The horizontal axis is plotted on a log scale.
OR, odds ratio; na, non-applicable.
The overall analysis investigating the association between null genotype of GSTM1 gene and the risk of developing CML relative to the normal genotype revealed nonsignificant heterogeneity between the studies (pQ = 0.87, I2 = 0%). The association was also not significant (OR = 1.00 [0.83-1.20]). In subgroup analysis, according to ethnicity, OR was nonsignificant in Whites, East Asians, and Indians: OR = 1.38 (0.43-4.46), 0.94 (0.65-1.35), and 1.16 (0.74-1.82), respectively.
The analysis of the GSTT1 gene and its association with CML revealed that the null versus normal genotype allele showed significant heterogeneity between studies (pQ = 0.04, I2 = 53%), the RE pooled OR being significant (OR = 1.57 [1.13-2.17]). In subgroup analysis, results for Indians were significant (OR = 2.89 [1.56-5.35]) and for East Asians were nonsignificant (OR = 1.07 [0.74-1.54]).
The analysis investigating the association between the combined GSTM1 null/GSTT1 null genotype and the risk of developing CML relative to the GSTM1 normal/GSTT1 normal genotype showed significant heterogeneity between the studies (pQ = 0.04, I2 = 63%). The RE OR was not significant (OR = 1.72 [0.68-4.33]). Nonsignificant results were also obtained for the GSTM1 null/GSTT1 normal genotype: OR = 0.93 (0.63-1.37), pQ = 0.45, I2 = 0%. However, the risk effect for the GSTM1 normal/GSTT1 null genotype showed a significant association and lack of heterogeneity: OR = 1.95 (1.17-3.24), pQ = 0.65, I2 = 0%.
Potential bias
Whether genotyping was blinded to clinical status was not disclosed in any of the studies. Cumulative meta-analysis for the GSTM1 gene showed that the ORs declined from 3.28 in 1999 (first study [Lemos et al.]) to 1.38 in 2001 and then remained fairly constant in the period 2005-2008 (Fig. 3). The association remained nonsignificant during the whole period. In recursive cumulative meta-analysis, the relative change in OR did not stabilize at a specific value (0.42 in 2001/1999, 0.72 in 2005/2001, 1.04 in 2007/2005, and 0.97 in 2008/2007), indicating that more evidence is needed to claim or deny an association.

Cumulative meta-analyses for the null vs. normal genotype contrast of GSTM1 and GSTT1 genes. RE OR with the corresponding 95% CI at the end of each year (information step) is shown. The horizontal axis is plotted on a log scale.
In cumulative meta-analysis for the GSTT1 gene, the odds increased monotonically from 1.34 in 2001 to 1.57 in 2008 (Fig. 3). However, the relative change in OR was not stable (1.12 in 2005/2001, 1.10 in 2007/2005, and 1.01 in 2008/2007), indicating the need for more evidence to support a definite association.
The Egger test for both the GSTM1 and GSTT1 genes revealed a differential magnitude of effect in large versus small studies (p = 0.03 and 0.04, respectively). In GSTT1, the studies with the smallest number of cases (Mondal et al., 2005; Bajpai et al., 2007; Taspinar et al., 2008) showed significant association whereas the larger studies (Löffler et al., 2001; Hishida et al., 2005; Lourenco et al., 2005; Chen et al., 2008; Souza et al., 2008) did not.
Discussion
The present meta-analysis examined variants in the GSTM1 and GSTT1 genes and their relationship with the risk of developing CML. The strength of this analysis lies in the accumulation of published data giving greater information to detect significant associations (Zintzaras and Lau, 2008a, 2008b). Usually in the meta-analysis of genetic association studies, the sample sizes of individual studies tend to be small and the power of single studies is very low. Synthesis of data from many studies is expected to improve power and the gain could be considerable, unless there is very large genuine heterogeneity between studies (Zintzaras, 2006b; Trikalinos et al., 2008; Zintzaras and Lau, 2008a). However, power calculations are usually considered inappropriate in meta-analysis since the data are already assembled (Zintzaras and Lau, 2008a).
Overall, the meta-analysis showed nonsignificant association with CML for GSTM1 gene and significant results for GSTT1. This association holds true especially in subjects of Indian and East Asian descent. The combined genotypes of GSTM1 and GSTT1 revealed association with risk of developing CML only for GSTM1 normal/GSTT1 null genotype, indicating that the risk effects differ depending on which gene possesses the null genotype. However, these results should be interpreted with caution as they are based on a limited number of studies.
Heterogeneity in the studies ranged from none (in GSTM1) to high (in GSTT1). High heterogeneity might be due to genuine differences in the studied populations and differences in study design and conduct. To avoid heterogeneity, future studies should incorporate other similar studies in a meta-analysis (Zintzaras and Lau, 2008a). In addition, meta-analysis offers the opportunity to place each study in the context of all others and to examine why studies reach different conclusions (Zintzaras et al., 2006, 2008).
The test of bias revealed a differential magnitude of effect in large versus small studies. In particular for GSTT1 gene, the studies with the smallest number of cases showed significant association and the large studies showed nonsignificant associations. For GSTM1 gene, the cumulative meta-analysis showed a downward trend in association; in contrast, GSTT1 gene showed an upward trend. However, in both genes there was instability in the relative change of risk effects, indicating that more evidence is needed to definitely claim or deny an association. In all studies the genotyping test used did not distinguish whether the GST normal genotype consists of one (heterozygotes) or both alleles (homozygotes). Thus, the quality of the studies in terms of the Hardy-Weinberg equilibrium cannot be tested, and genotyping errors and/or population structure may exist in the individual studies (Zintzaras and Lau, 2008a).
The overall lack of association for GSTM1 gene and the discrepant results for GSTT1 might be due to other variants involved in phase II metabolic pathway or unidentified functional mutations in the GST genes that affect the risk of developing CML. Gene-gene interactions can be major determinants of CML risk rather than the individual variants, and a meta-analysis of genotype combinations or haplotypes may provide more reliable information than single gene variants (Zintzaras et al., 2006; Zintzaras and Lau, 2008a). The search for susceptibility loci can be complicated by the increased number of contributing loci and susceptibility alleles (Zintzaras and Ioannidis, 2008). Then elucidation of the pathogenesis of CML may demand the examination of a large number of genetic variants in the same or different pathophysiological pathways (Kitsios and Zintzaras, 2007; Zdoukopoulos and Zintzaras, 2008). However, there is some evidence that combination of GSTM1 and GSTT1 null genotypes confers a high risk of cancer (Bajpai et al., 2007). GSTM1 and GSTT1 genes have already been implicated in other types of cancer such as chronic lymphocytic leukemia, breast cancer, gastric cancer, lung cancer, and colorectal cancer, and therefore their role in CML development cannot be excluded (Vogl et al., 2004; Chen et al., 2005; Saadat, 2006; Hosgood et al., 2007; Zintzaras and Kitsios, 2009).
In addition to candidate gene studies, microarray gene expression analyses (Zintzaras and Ioannidis, 2008) and gene-wide association studies may assist in the selection of candidate polymorphisms by identifying the most active genes involved in the disease pathophysiology (Zintzaras and Lau, 2008a). A possible genomic convergence of the different data sources may provide further insights into the mechanisms involved in CML (Kitsios and Zintzaras, 2009).
Other complex mechanisms and environmental interactions are expected to play a crucial role in the pathogenesis of CML (Chen et al., 2008). There are known environmental factors that may be related to increased risk of CML, such as exposure to benzene and radiation; however, the existence of unknown factors cannot be excluded. Despite difficulties in study design and assessment of the exposures, such factors should be incorporated in future studies and meta-analyses (Zintzaras and Lau, 2008a).
In summary, the accumulated evidence indicated an association between GSTT1 gene and CML and large heterogeneity between studies. The association for GSTM1 was nonsignificant. However, the results of the present meta-analysis were based on relatively small numbers of studies and participants, and consequently their interpretation has to be cautious (Zintzaras, 2006a; Zintzaras and Lau, 2008a). Long-term prospective and case-control studies (Zintzaras and Lau, 2008a) investigating gene-gene and gene-environment interactions and the use of current genomic data (Zintzaras, 2006a; Zintzaras and Ioannidis, 2008) may provide more conclusive claims about the genetics of CML.
Footnotes
Disclosure Statement
No competing financial interests exist.