
Abstract
Toll-like receptor 2 (TLR2) is a recognition receptor for the widest repertoire of pathogen-associated molecular patterns. Two polymorphisms of TLR2 could be linked to reduced nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) activation and to increased risk of infection (supposed-2029C>T and 2258G>A). We investigated the supposed-2029C>T and 2258G>A TLR2 polymorphisms in 422 critically ill patients of European origin from southern Brazil (295 with sepsis and 127 without sepsis) and reviewed 33 studies on these polymorphisms, conducting a quality assessment with a score system. Among our patients we found only one heterozygote (1/422) for the supposed-2029C>T and none for the 2258G>A (0/422) single nucleotide polymorphism (SNP). We were unable to find a clinical application of supposed-2029T and 2258A allele analyses in our southern Brazilian population. Our review detected that current TLR2 SNP assays had very controversial and contradictory results derived from reports with a variety of investigation quality criteria. We suggest that, if analyzed alone, the supposed-2029C>T and 2258G>A TLR2 SNP are not good candidates for genetic markers in studies that search for direct or indirect clinical applications between genotype and phenotype. Future efforts to improve the knowledge and to provide other simultaneous genetic markers might reveal a more effective TLR2 effect on the susceptibility to infectious diseases.
Introduction
T
TLR2 (Reactome-UniProt: O60603) has a special place among the 10 members of the human TR family. Of all mammalian TLRs and, perhaps, of all pathogen recognition receptors, TLR2 is capable of detecting the widest repertoire of pathogen-associated molecular patterns (Texereau et al., 2005). The variety of microbial components that TLR2 recognizes are lipoproteins from gram-negative bacteria, Mycoplasma, and spirochetes, peptidoglycan and lipoteichoic acid from gram-positive bacteria, lipoarabinomannan from mycobacteria, glycoinositolphospholipids from Trypanosoma cruzi, modulin from Staphylococcus epidermidis, zymosan from fungi, glycolipids from Treponema maltophilum, and porins that constitute the outer membrane of Neisseria (Takeda et al., 2003). One aspect of TLR2 ligand recognition involves cooperation with other TLR family members, particularly with TLR6 and TLR1, for discrimination among different microbial components (Takeda et al., 2003). To enable the innate system to recognize numerous structures of lipopeptides in various pathogens, distinct acylation patterns are recognized by either TLR2/TLR1 or TLR2/TLR6, the former heterodimer only recognizes triacylated lipopeptides, whereas the latter recognizes diacylated lipopeptides. There is evidence that the charge of the amino acids, especially the charge of the C-terminal amino acids, may determine the signaling through TLR2/TLR1 or TLR2/TLR6 (Buwitt-Beckmann et al., 2006). Reactome database (Reactome, 2008; www.reactome.org/) demonstrates that these two triggers release the same activation cascade through MyD88.
Only two of the nonsynonymous single nucleotide polymorphisms (SNP) (one of these is just a supposed polymorphism) of the cytoplasmic domain of TLR2 have been linked to reduced nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) activation and to increased infection risk, confirming animal models that suggest that defective TLR2 signaling is a causative factor for increased susceptibility to bacterial disease (Texereau et al., 2005). The first is the suggested C>T substitution at position 2029 from the start codon. Located in exon 3, this 2029C>T SNP results in replacement of a conserved arginine residue by tryptophan at position 677 (Arg677Trp protein mutation). As this arginine residue is highly conserved at the C-terminal of the TLR family, its mutation is likely to affect the molecule signaling function (Kang and Chae, 2001). Nevertheless, Malhotra et al. (2005) proposes that it is not a true polymorphism because a C is fixed at the authentic (functional) TLR2 exon 3 sequence, but a T occupies the same position in a pseudo-exon 3 upstream to the TLR2 gene. Both regions (exon 3 and pseudo-exon 3) are simultaneously recognized by primers, and the double amplification is indicative of an erroneous 2029C>T SNP suggestion and an apparent heterozygosis in the populations.
The second, and more proper, SNP is a G>A change at 2258 from the start codon of the TLR2 gene (2258G>A; rs5743708), which causes an arginine to glutamine substitution at residue 753 (Arg753Gln). As this arginine is conserved between mice and humans and is part of a highly conserved stretch of amino acids at the C-terminus of TLR2, its change would have important phenotypic effects (Lorenz et al., 2000). The HapMap (2008) shows the 2258G>A nucleotide substitution as polymorphic just in the European Caucasian population, with a 5% frequency of the 2258A allele, with 2258G fixed in the oriental and black populations.
Based on the above, it would be expected that polymorphic alterations in the TLR2-codifying sequence could cause a hyporesponsive TLR2 pathway and affect susceptibility to severe infections. Thus, we tested whether the supposed-2029T and 2258A rare alleles of TLR2 would be outnumbered in patients with sepsis when compared with matching individuals without sepsis in a well-characterized population of 422 white, critically ill patients. We also performed a detailed review on TLR2 and a discussion about the controversial nature of these mutations on different diseases and populations, to analyze whether identification of the supposed-2029T and 2258A rare alleles would have clinical applications.
Materials and Methods
Study on patients
Design, subjects, and approval
This single-center, observational, retrospective, cohort study was conducted with data from random patients admitted to the intensive care unit (ICU) of the São Lucas Hospital (HSL), Brazil, between January 1, 2004 and December 31, 2006. The ICU-HSL is a general nonpediatric Medical-Surgery Intensive Care Unit with 13 beds, which receives about 300-400 patients/year. We worked on the archived DNA collection from septic and nonseptic ICU patients (controls). We monitored patients daily during their entire ICU and post-ICU (hospital) stay. Patients were not eligible if they were diagnosed with HIV infection, with known immunodeficiency, taking immunosuppressive drugs, pregnant, or lactating. A total of 422 white, critically ill adult patients from southern Brazil (225 male and 197 female patients) admitted to the ICU were included in this study. We also studied 92 random healthy DNA donors from the Paternity Investigation Unit who served as the population stratification group. All subjects were from southern Brazil, which is composed by a singular genetic background: a majority of subjects with European origin (Portuguese, Italian, Spanish, and German ancestry) and a small amount of individuals with African traits contributing to their genetic pool (Parra et al., 2003). This sepsis-genotyping project was approved by the Research Ethics Committee of the Pontifical Catholic University of Rio Grande do Sul (protocols 03-01732 and 07-1500; REC Tel.: 55 51 3320 3345), and informed written consent or assent to participate was obtained from all subjects or patients' surrogates.
Phenotyping
The patients admitted in the general nonpediatric Medical-Surgery ICU of the HSL were diagnosed for sepsis and sepsis-related conditions (severe sepsis and septic shock) according to the American College of Chest Physicians/Society of Critical Care Consensus Conference definition (ACCP/SCCM, 1992). Sepsis was defined as systemic inflammation, caused by infection, or occurring in the presence of clinical evidence of infection: septic patients were diagnosed with, at least, one infection focus, or had clinical evidence of infection, and were treated with wide-spectrum antibiotics. Systemic inflammation (systemic inflammatory response syndrome) was defined by the presence of at least two of the following symptoms: fever or hypothermia (temperature in the core of body >38°C or <36°C); tachycardia (ventricular rate >90 heartbeats/min); tachypnea or hyperventilation (breaths/min >20 or PaCO2 <32 mmHg); leucocytosis or leucopenia. If these symptoms were complicated by organ dysfunction, the definition of severe sepsis was met. As all our subjects were at ICU with some organ dysfunction, however, we called them patients with sepsis. If persistent arterial hypotension was present, the term “septic shock” was applied. The sepsis and septic shock patients' diagnoses were attributed by ICU physicians during the permanence of patients in ICU.
For illness severity evaluation we used the acute physiology and chronic health evaluation II (APACHE-II) score (Knaus et al., 1985) obtained on ICU admission day and used as an estimate for severity of disease. For organ dysfunction evaluation we used the sequential organ failure assessment (SOFA) (Vincent et al., 1998) score obtained on ICU admission day (SOFA-1) and daily during the first week from the ICU admission and on days 15 (SOFA-15) and 29 (SOFA-29) for patients who stayed in the ICU. Temporal variation comprised length of stay in ICU and ICU plus post-ICU (hospital) stay. Mortality was measured in days until death in total hospital stay: clinical endpoints of the study were discharge from the hospital (considered survivors) or death (considered nonsurvivors). For those patients with multiple ICU admission during the study period, only data from the first entrance was considered. All clinical data were collected and verified by ICU physicians with controls.
Genotyping
Genomic DNA was isolated from leucocytes by standard procedures and maintained at −20°C (Lahiri and Nurnberger, 1991). A 340-bp PCR product was designed to encompass both SNPs under study using forward primer TLR2-F 5′-GCC-TAC-TGG-GTG-GAG-AAC-CT-3′ and reverse primer TLR2-R 5′-GGC-CAC-TCC-AGG-TAG-GTC-TT-3′ (Invitrogen-Life Technologies, São Paulo, SP, Brazil). In this genotyping, we applied primers described by Schröder et al. (2003) that are different than the PP1-primer reported by Malhotra et al. (2005), which amplified both exon 3 and pseudo-exon 3, identifying the presence of supposed-2029T nucleotide in 100% of individuals. Each reaction was performed in 25 μL containing 10-50 ng DNA, 0.12 μM each primer, 0.2 mM deoxynucleotide triphosphate, 1.5 mM MgCl2, and 1 U Taq polymerase in Taq 1× Buffer (LGC Biotecnologia, Cotia, Brazil). The reactions were made in a TC-412 thermocycler (Barloworld Scientific Ltd., Stone, Staffordshire, United Kingdom) as follows: 95°C for 10 min; 35 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s; and 72°C for 5 min for final extension. The amplified PCR products were genotyped by the method of direct sequencing (BigDye TerminatorCycle Sequencing Kit and ABI PRISM 3100 Avant Genetic Analyzer; Perkin-Elmer, Foster City, CA) using the forward primer, and the results were confirmed by sequencing with reverse primer. At least 10% of the samples were subjected to a second, independent PCR-sequencing reanalysis cycle to confirm the genotypes. To confirm that the 340-bp PCR-amplified product really represented the targeted product, we performed a latest sequence analysis in a MegaBase 1000 capillary DNA sequencer (Amersham Biosciences UK Ltd., Chalfont St. Giles, Bucks, United Kingdom), also using the designed primers (forward and reverse). Each sequence obtained was submitted to an online BLASTn alignment (BLAST, 2008; www.ncbi.nlm.nih.gov/BLAST/), and we found consensus with the Homo sapiens TLR2 gene, promoter region (GenBank accession NM_003264), and the sequence exported from chromatogram file. The alignment view was performed in ClustalX program [version 1.8, as described by Thompson et al. (1997)] in multiple alignment modes, with sequences loaded in FASTA format. Blank control wells were always used to test contamination of the PCR reagents. All the personnel involved in patient care were blind to the selection process and genotyping results.
To have information about our population frequencies, we obtained genotypic and allelic frequencies from healthy controls. We noticed that supposed-2029C and 2258G alleles were fixed in the healthy population. We did not use healthy subjects as control group because we assumed that the environmental exposure has a crucial influence, and therefore, we performed comparison among ICU patients.
Assessment of the review
Search strategy
A literature review was performed in the PubMed database covering all papers published from 1998 to 2008. Search terms were a combination of the following keywords: TLR2, polymorphism, 2029C>T, 2258G>A, Arg677Trp, and/or Arg753Gln. Additionally, the reference lists of selected papers were used to identify studies that were not collected in the database. We included articles that were available in English, and studies that reported disease and/or association with the TLR2 polymorphisms were also included (Clark and Baudoin, 2006; Flores et al., 2008; Geng et al., 2008; Shi et al., 2008).
Study quality assessment
We used a five-point scoring classification based on previous criteria adopted from published recommendations on the assessment of the quality of genetic association studies. The criteria and scores are described in detail below; we scored as 1 if present, or 0 if absent (Clark and Baudoin, 2006). These independent criteria were included in Figure 1, and the total score (0-5) was applied in Tables 1 and 2.
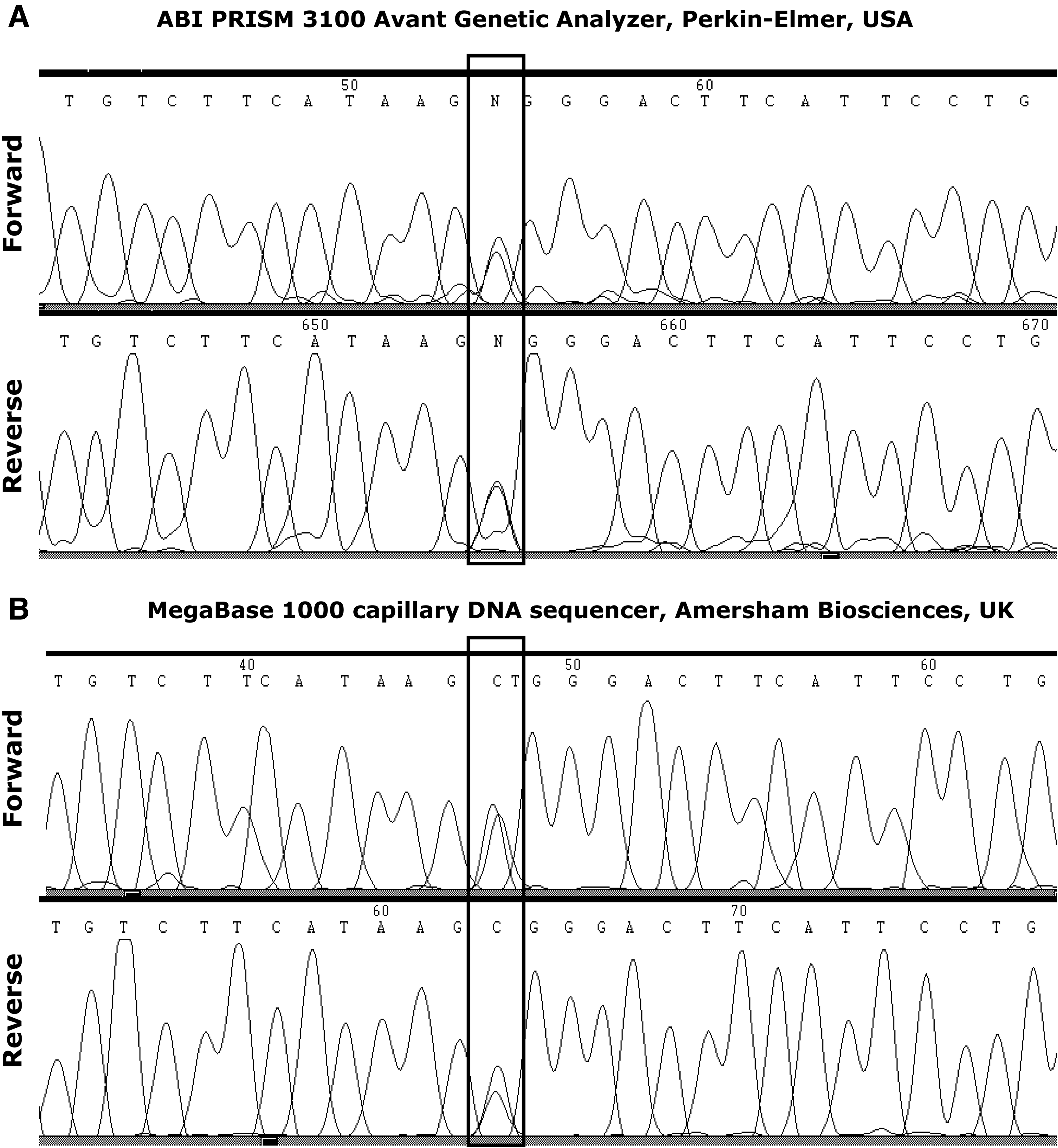
Genotyping analysis of the supposed-2029C>T toll-like receptor 2 (TLR2) single nucleotide polymorphism (SNP) (into the box) via sequencing in an ABI PRISM 3100 Avant Genetic Analyzer (Perkin-Elmer, Foster City, CA) (
p-Value describes a comparison between patients with sepsis and without sepsis.
n (%).
Median (IQR).
Mean (SD).
IQR, interquartile range; SD, standard deviation of the mean; ST, Student's t-test; MW, Mann-Whitney U-test; ×2, Pearson chi-square test; LOS, length of stay; HW, Pearson chi-square test for Hardy-Weinberg equilibrium; APACHE-II, acute physiology and chronic health evaluation II; SOFA, sequential organ failure assessment; ICU, intensive care unit; TLR2, toll-like receptor 2.
No association study.
S, five-point scoring classes; HW Eq., Hardy-Weinberg Equilibrium; OR, odds ratio; CI, confidence interval; ns, not significant; NI, not informed; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; NTM, nontuberculous mycobacterial; RFLP, restriction fragment length polymorphism; SSCP, single-strand conformation polymorphism.
Control group
Score 1: When the control group was fully described and/or the work was clear if more than one group was used or if control data were reused from a previous study (Bird et al., 2001; Clark and Baudoin, 2006). Score 0: When the control group was absent.
Hardy-Weinberg equilibrium
Score 1: When the Hardy-Weinberg equilibrium was tested (the Hardy-Weinberg equilibrium is a state in which allele frequencies in a population tend to remain the same from generation to generation unless acted on by outside influences) (Hattersley and McCarthy, 2005; Clark and Baudoin, 2006). In this category we also discriminated between studies in which the sample was in Hardy-Weinberg equilibrium or not. Score 0: When the Hardy-Weinberg equilibrium test was not cited.
Blinding
Score 1: When the study had cited that genotyping was blind to the clinical status of the patient (Clark and Baudoin, 2006). Score 0: When the blinding strategy was not cited.
Control for confounders
Score 1: When the study tests were adjusted to confounding variables and/or multiple comparisons were executed to test for more than one association in this study and/or the study tests were repeated in an independent group of cases and controls (Vitali and Randolph, 2005). Score 0: When the control for confounders was not used.
Duplicated genotyping
Score 1: When duplicate genotyping was performed on the same assay or on another assay (Hattersley and McCarthy, 2005). Score 0: When duplicate genotyping was not performed.
Frequency calculation and presence of association
The allelic frequencies of both SNPs studied were calculated based on the following formula: the number of rare alleles divided for the total number of alleles, for each polymorphism. We considered as statistically significant the association between genotype/allele and phenotype when the study presented a p < 0.05 in, at least, a nonadjusted analysis.
Results
Study on patients
Table 1 shows clinical and demographic data of our sample of critically ill patients. Four hundred and twenty-two patients were investigated: data about age, sex, APACHE-II and SOFA scores, length of stay in ICU and ICU plus hospital, septic shock occurrence, cause of admission, and mortality rates are shown for patients with or without sepsis. The incidence of sepsis in ICU patients was 69.9% (295/422), 71.5% of which had septic shock (211/295). The frequency of septic shock in ICU patients was 50.0% (211/422). Sepsis was positively associated with older age, higher APACHE-II and SOFA-1 scores, longer ICU stay, and medical admission causes (all p < 0.01). Accordingly, mortality rates were higher in septic than nonseptic patients (p < 0.001).
The main causes of admission to ICU were medical sepsis (35%; 147/420), followed by medical respiratory (24.5%; 103/420) and surgical abdominal (11.4%; 48/420). Microbiological results from blood sample of patients with sepsis (n = 295) were as follows: 55.1% (162/295) with no focus identified; 21.8% (64/295) with only gram-negative bacteria; 6.8% (20/295) with only gram-positive bacteria; 9.9% (29/295) with both gram-negative and gram-positive bacteria; 1.4% (4/295) with fungal infection; and 5.1% (15/295) with fungal and bacterial infection. The anatomical distribution of the primary site of infection in septic patients was as follows: 65.7% (194/295) pulmonary; 22.8% (67/295) abdominal; 4.1% (12/295) urinary; 1.7% (5/295) central nervous system; 1.7% (5/295) skin; and 2% (6/295) from another site.
The genotypic and allelic frequencies in our sample to supposed-2029C>T and 2258G>T SNPs were as follows: 2029CC = 0.998 (421/422), 2029CT = 0.002 (1/422), and 2029TT = 0; 2029C = 0.99 (843/844) and 2029T = 0.01 (1/844); 2258GG = 1 (422/422), 2258GA and 2258AA = 0; 2258G = 1 (844/844); 2258A = 0. The phenotypes (sepsis, septic shock, organ dysfunction, or mortality) were not related to genotypes because of the very low frequency of the supposed-2029T allele and null frequency of the 2258A allele. The frequencies apparently did not differ from values expected by the Hardy-Weinberg model. As only one mutant allele was identified to supposed-2029C>T SNP in our sample, we show the amplified sequences to subsequent potential analysis (Fig. 1).
Assessment of the review
We have compiled 33 reports published from 1998 to 2008 regarding the supposed-2029C>T and/or 2258G>A TLR2 SNPs. The TLR2 SNP investigations have grown up in the last 10 years (Fig. 2), especially in 2007 when 27.8% (5/18) and 39.3% (11/28) of the reports on the supposed-2029C>T and 2258G>A SNPs, respectively, were published. Tables 2 and 3 present our compilation with the main characteristics and findings of each study, as follows: methods, population, number of cases and controls, frequencies of genotypes and alleles, and the study quality scores. In the last columns are noted the end points and the statistical data of association tests (when applicable).
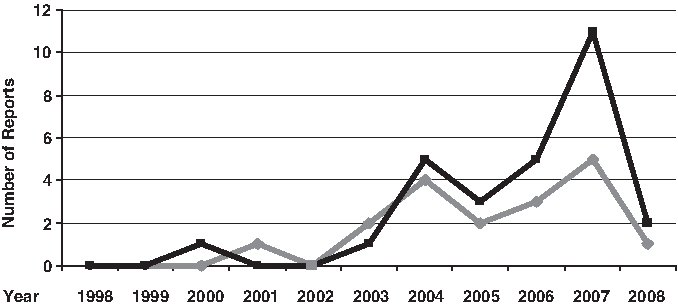
Distribution of TLR2 studies by time. Number of studies with supposed-2029C>T (gray line) and 2258G>A (black line) single nucleotide polymorphism (SNP), from 1998 to 2008.
No association study.
Calculation was not possible.
S, 5-point scoring classes; HW Eq., Hardy-Weinberg equilibrium; OR, odds ratio; CI, confidence interval; ns, not significant; NI, not informed; UK, United Kingdom; USA, United States of America; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; NTM, nontuberculous mycobacterial; CAIMT, carotid artery intima-media thickness; DPMD, different pattern microbial defense; PTCA, percutaneous transluminal coronary angioplasty.
The majority of TLR2 SNP studies was concentrated in three regions of the world: Europe (45.4%; 15/33) (mainly in Germany), Turkey (24.2%; 8/33), and South Korea (12.1%; 4/33). A few other reports originated from the United States (6.0%; 2/33), Colombia, India, Tunisia, and Taiwan (all 3.0%; 1/33 each).
The supposed-2029T allele was null (0%) in Europe, but in Tunisia it was found in 47% of cases and 15% of control subjects (Ben-Ali et al., 2004), and in Korea, it was observed in 6% and later in 17% of case individuals (Kang and Chae, 2001; Kang et al., 2004). The frequency of 2258A allele was from 1% to 9% in Europe and it was higher in three reports from Turkey [14% (Ogus et al., 2004), 23% (Kutukculer et al., 2007), and 46% (Berdeli et al., 2005)]. In the two studies from the United States, 2258A allele frequency was reported as 9% (Eid et al., 2007; Kijpittayarit et al., 2007) and in a report from Colombia both mutant alleles were null (Zafra et al., 2008). To better visualize the TLR2 mutant allele frequencies around the word, we showed it in the maps (Fig. 3).
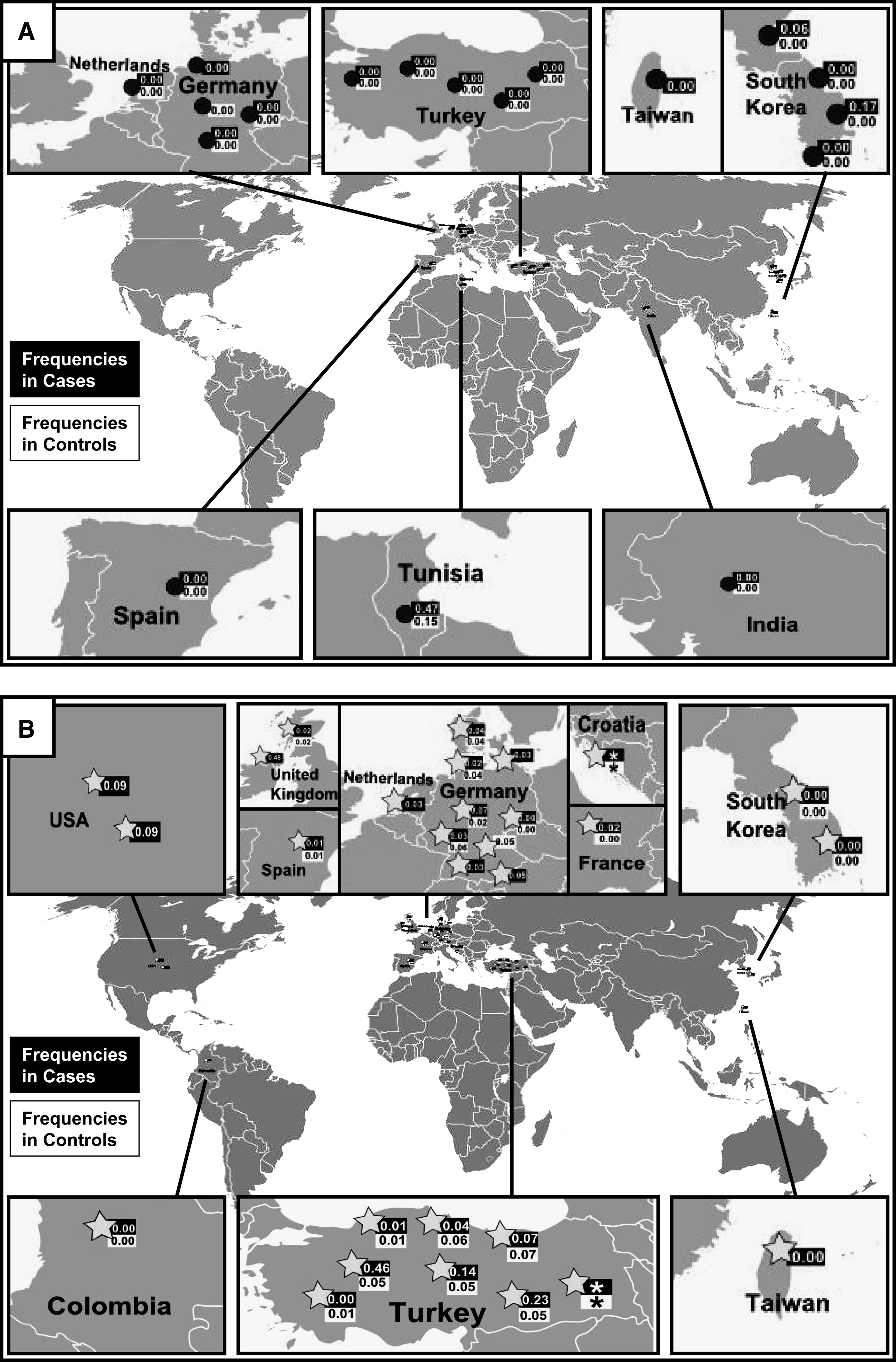
Distribution and frequencies of TLR2 supposed-2029C>T (
Most reports revealed no statistically significant association between mutant alleles and phenotypes: 16.7% (3/18) to supposed-2029C>T and 42.8% (12/28) to 2258G>A SNP. All studies (100%; 3/3) to supposed-2029C>T and 41.7% (5/12) to 2258G>A SNP were performed with less than 200 subjects (Fig. 4). We detected four works (1/3 for supposed-2029C>T and 3/12 for 2258G>A SNPs) that report a statistically significant association but did not cite the p-value (Tables 2 and 3).

Sample size in TLR2 2029C>T (gray bars) and 2258G>A (black bars) reports. Striped box inside each bar represents the percent of studies that observed positive association with mutant alleles; where there is no striped box the percent is zero.
None of the set of 33 TLR2 SNP reports covered the five points to scoring classes; only 11.1% (2/18) to supposed-2029C>T and 6.9% (2/28) to 2258G>A SNP comprised four points (i.e., in these works, four of five points were present), but a majority of the reports considered two of the five-point scoring classes (Fig. 5). Figure 6 shows a detailed comparison among all studies according to the five-point scoring classes to quality assessment. A control group was included in 94.4% (17/18) of the studies concerning the supposed-2029C>T and 78.6% (22/28) to 2258G>A SNP studies. Around 22% (4/18) to supposed-2029C>T and 35% (10/28) to 2258G>A of investigations mentioned the Hardy-Weinberg equilibrium testing. The blinding strategy was reported in only three of studies (9.1%; 3/33). Only 11.1% (2/18) to supposed-2029C>T and 28.6% (8/28) to 2258G>A performed the adjusted analysis of confounding variables, and around 60% (11/18) of supposed-2029C>T and 40% (11/28) of 2258G>A achieved the standard for reproducibility with a second confirmatory assay (Fig. 6).
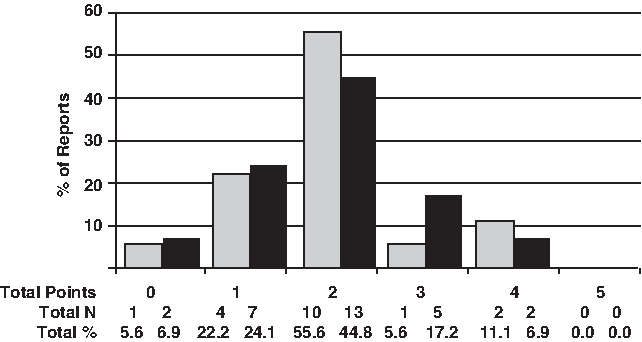
Quality criterion points in TLR2 2029C>T (gray bars) and 2258G>A (black bars) reports. For description of quality criterion points and scores, see Materials and Methods section.
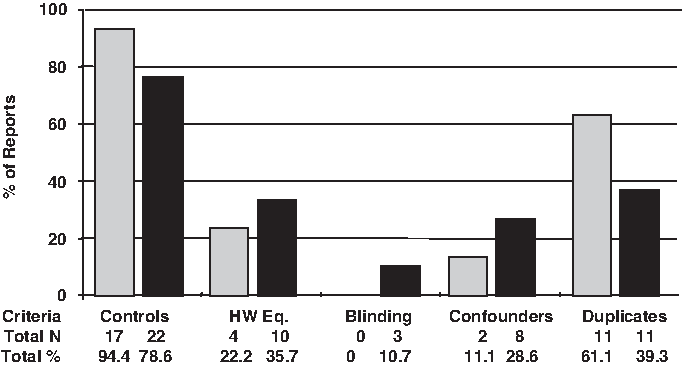
Quality criterion categories in TLR2 2029C>T (gray bars) and 2258G>A (black bars) reports. For description of quality criterion categories and scores, see Materials and Methods section. HW Eq., Hardy-Weinberg equilibrium.
In both SNP analyses the most used laboratory method for genotyping (around 65%) was PCR, followed by restriction fragment length polymorphism (RFLP) examination (Fig. 7). When using this PCR-RFLP technique it is recommended to perform duplicate assays (with the same or other method) to detect discrepancy between results (Hattersley and McCarthy, 2005), and ideally, when >10% of genotyped samples are inconsistent the assays should be redone. We noticed that the duplicate strategy was employed in 75% (to supposed-2029C>T SNP) and 52.6% (to 2258G>A SNP) of the PCR+RFLP assays performed (Fig. 7).
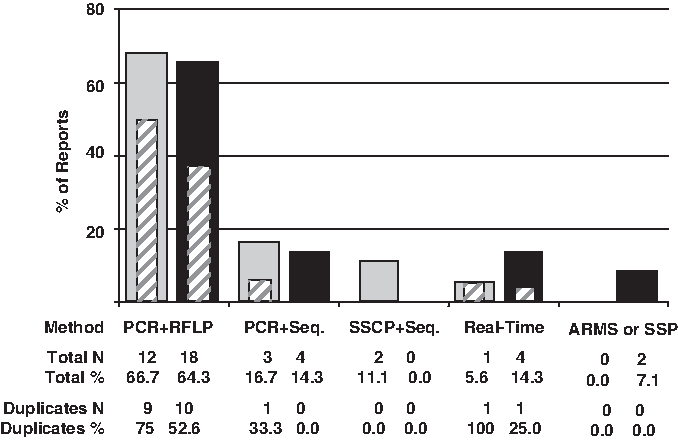
Methods used to perform TLR2 2029C>T (gray bars) and 2258G>A (black bars) genotyping. Striped box inside each bar represents the percent of studies that have duplicated analysis; where there is no striped box the percents is zero. PCR, polymerase chain reaction; RFLP, restriction fragment length polymorphism; SSCP, single-strand conformation polymorphism; Seq., automatic sequencing by Sanger or enzymatic method (pyrosequencing); Real-time, real-time PCR; ARMS, amplification refractory mutation system; SSP, sequence specific primer.
Discussion
It is well known that TLR2 is an important cellular receptor responsible for the recognition of a large range of pathogens and activation of inflammatory cytokines production, having an essential role in the innate and acquired immunity. Polymorphic mutations on its codifying sequence could produce a hyporesponsive TLR2 pathway and affect susceptibility to infections. Thus, investigations about TLR2 mutant alleles have a biological plausibility, because of a possible causal effect for TLR2 in the pathogenesis of the infectious diseases. As the most studied TLR2 SNPs have been the supposed-2029C>T and the 2258G>A, we investigated these two polymorphisms in 422 critically ill patients to test whether the mutant alleles would affect the critically ill patients' susceptibility or outcome and we found that these alleles are rare (supposed-2029T) or null (2258A) in our population.
To discuss our results and to inquire about their potential clinical applications, we performed a review with 33 reports. We observed that TLR2 SNP investigations have grown up, especially in 2007, and they were basically distributed in three regions around the world: Europe, Turkey, and South Korea, with the American and African continents with just a few studies. In face of these few reports, we assume that the literature do not expose the actual repertoire of TLR2 genotyping performed worldwide, because essays concerning null or rare alleles are more likely to remain unpublished. Additionally, failure in detecting significant association may have inhibited new investigations.
Initial studies usually yield suggestive rather conclusive results because they are likely to overestimate the true effect size (Hattersley and McCarthy, 2005). Although an adequate sample size and power calculations are necessary to assure conclusions and exclude false-negative results, this was a rare finding among the TLR2 reports analyzed: the TLR2 SNP studies did not mention the power calculation to establish the sample size. The concept of power is closely related to the two types of statistical errors: the type I error, named α (i.e., the probability of rejecting a true hypothesis), and the type II error, named β (i.e., the probability of accepting a false hypothesis). Power is defined as 1-β, that is, the probability of rejecting a false hypothesis, or the probability of not making a type II error (Sluis et al., 2008). The basic aim of a power study is to determine the sample size N, which is required to achieve adequate power, given a chosen α and a particular effect size. Regarding a hypothetic very high frequency of mutant TLR2 alleles (case: mutant allele = 0.20; control: mutant allele = 0.01) in a case-control study, we calculated that at least 10,000 subjects would be necessary to achieve 80% power to obtain a p-value of 0.05 in studies involving infectious diseases. Therefore, because of the impossibility of obtaining such a large sample, we recognize the importance of small sample size investigations as they are pioneers and further systematic meta-analysis could be evaluating the impact of these individual reports.
Approximately 35% of the 33 TLR2 papers had less than 200 subjects, around 50% of which had positive associations between mutant allele and phenotype. All studies with supposed-2029C>T SNP and more than 200 subjects (61%; 11/18) had no statistically significant association. In addition, even in that 43% (12/28) of the 2258G>A studies that had met the parameters for significant association, the largest study (with >600 subjects) showed no association. A study with more than 600 subjects tends to be more robust than one with less, and an inefficient sampling strategy can be compensated by an increase in sample size. Although there are some possible problems with very large samples (as the misclassification of outcome or the population heterogeneity), we would like to warn about the risk that studies with <200 finding an association have incurred type II error.
Altogether, it is noteworthy that most studies (83%; 15/18) did not find any association between the supposed-2029C>T TLR2 SNP and illnesses, probably because of the concurrence of sample size or the rarity of this allele. Nonetheless, Ben-Ali et al. (2004) exposed a strong positive association with tuberculosis (p < 0.0001), and Kang and Chae (2001) with lepromatous leprosy (without p-value). Similar to Kang and Chae's (2001) work, three other reports assured significant association despite not showing statistical parameters (Lorenz et al., 2000; Kang and Chae, 2001; Merx et al., 2007; Woehrle et al., 2008), and these would be essential information in association studies because they confirm results and warrant credibility to the data. Statistical imprecision, such as the absence of the p-value and power calculation, lack of Hardy-Weinberg equilibrium testing, or multiple statistical analyses, makes a pool of inaccurate TLR2 SNP studies.
Although the supposed-2029T allele is rare in many world populations (0% in Europe), in Tunisia, Ben-Ali et al. (2004) found this allele to be present in 47% of cases and 15% of controls, and in Korea, Kang et al. found it in 6% and later in 17% of their cases (Kang and Chae, 2001; Kang et al., 2004). It is possible that the frequency of this SNP will be altered in small populations or in subgroups of particular populations (small, culturally isolated communities that are mostly closed breeding groups) that have been exposed to founder effect (occurs when a small amount of people have many descendants surviving after a number of generations) or to specific environmental or lifestyle factor pressures. Interbreeding causes a random change in genotypic frequencies, particularly if the population is very small (if the population is small, Hardy-Weinberg may be violated). In such cases, the frequency of an allele may begin to drift toward higher or lower values. The result for a population is often high frequencies of specific alleles inherited from the few common ancestors who first had them. Genetic drift produces changes, but there is no guarantee that the novel population will be more fit than the original one. Changes by drift are aimless and not adaptive, and thus a possible explanation why the supposed-2029T allele, which causes susceptibility to infectious diseases, is increased in Tunisia and Korea. Additionally, if the supposed-2029T allele was a protective allele, its higher frequency would reflect that selective pressures have favored the transmission of this allele in their antecedents, and it would be expected to be more common in specific populations. Although this is a possible explanation, we cannot discard the likelihood of laboratorial inaccuracy. Clark and Baudoin (2006) showed that a combination of methodological and analytical problems is likely to explain the failure to replicate results: technical troubles with the methods of identifying the mutant allele, for example, may lead to false-positive and inconsistent conclusions.
The most used laboratory method for TLR2 genotyping was the nonautomated PCR-RFLP technique, which has some advantages such as being cheap and does not requiring sophisticated equipment. However, when compared with other methods, it needs intensive work and the results can be delayed and inexact (Table 4). Automated sequencing and real-time PCR have elevated cost and require highly trained technicians and support, but provide advanced precision and sensitivity, and smaller contamination risk because they have minimized manipulation. Even so, automated sequencing and real-time PCR were used in fewer instances.
Regarding technical inconsistencies, Malhotra et al. (2005) advised that 2029-C>T is not a true polymorphism because the authentic TLR2 exon 3 sequence and a mutant pseudo-exon 3 upstream of TLR2 gene are simultaneously recognized by primers, and the double amplification generates the erroneous 2029C>T SNP, suggesting an apparent heterozygosis in the populations. In our genotyping, we applied primers described by Schröder et al. (2003) that are different from the PP1-primer reported by Malhotra et al. (2005), which amplified both exon 3 and pseudo-exon 3, identifying in 100% of individuals the presence of supposed-2029T nucleotide. With our primers we tried not to amplify the pseudo-exon 3, but we found (confirming by two different sequencing systems) one supposed-2029T allele in a unique heterozygote patient (Fig. 1). This could reveal that either the primer sequences of Schröder et al. (2003) recognized the pseudo-exon 3 or that the supposed-2029C>T SNP really exists. It is important to note that we studied a population with European origin (Caucasian), and the sample of Malhotra et al. (2005) was from India.
In a unique previous study from Colombia, both mutant alleles are null (Zafra et al., 2008). In Europe, the 2258A allele frequency ranged from 1% to 9% but, interestingly, it was higher in three singular reports from Turkey [14% (Ogus et al., 2004), 23% (Kutukculer et al., 2007), and 46% (Berdeli et al., 2005)]. Once more, apart from experimental or statistical inaccuracy, it would be possible due to different genetic backgrounds in these populations or singular patterns of lifestyle risk factors. Other flaws were also detected in 2258G>A TLR2 SNP reports. In one paper there is a mistake about the 2258G>A SNP (Arg753Gln) description: it changed the G allele to T allele (as 2258T>A) and in a following moment it changed the A allele to C (as 2258C>T) (Labrum et al., 2007). It was a small error but it can be a source of additional technical and interpretation mistakes.
In general, the set of 33 TLR2 reports examined presented some limitations such as the lack of well-designed strategies for analysis, which precluded precise significant final conclusions. In summary, our genotyping results do not support a clinical application for the supposed-2029T and 2258A alleles in the southern Brazilian population. Our review detected that current TLR2 SNP essays had very controversial and contradictory results derived from reports with a variety of quality criteria of investigation. We suggest that, if analyzed alone, the supposed-2029C>T and 2258G>A TLR2 SNPs are not good candidates for genetic markers in studies that search for direct clinical applications. Future efforts to improve the knowledge and to provide other simultaneous genetic markers might reveal a more effective TLR2 effect on the susceptibility to infectious diseases.
Footnotes
Acknowledgments
The authors thank C.A.S. Ferreira, C.L. Dornelles, and F.B. Nunes for their suggestions and D.D. Paskulin, T.J. Borges, C.O. Alminhana, C. Froes, and T.F.C. Silva for technical assistance. This study was financed by the Conselho Nacional de Desenvolvimento Científico e Tecnológico-CNPq (process no. 505536/2002-8), the Programa de Bolsa Pesquisa para Alunos da Graduação-BPA PUCRS 2007-2008, and Faculdade de Biociências, PUCRS. The study is part of the Masters' Degree dissertation of the first author.
Disclosure Statement
No competing financial interests exist.