
Abstract
Context: Conventional karyotyping for antenatal diagnosis is time consuming and hence there has been a growing interest in more rapid techniques for detection of chromosomal aneuploidies. Around 95% of Down syndrome cases are due to free trisomy 21. Aims: The aims of this study were to demonstrate sensitivity of DNA diagnosis of Down syndrome using polymerase chain reaction (PCR) and short tandem repeat (STR) markers, and to determine the parental origin of the nondisjoined chromosome. Methods: DNA polymorphism was studied using two tetranucleotide STR markers, D21S2055 situated at 21q22.2 and D21S11 situated at 21q21.1. PCR conditions for both markers were standardized. PCR products were analyzed in 15% poly-acrylamide gels. The results obtained by STR analysis were verified with the chromosomal analysis and quantitative fluorescent (QF)-PCR. Results: These two STR markers were able to detect 86.7% cases of trisomy 21. Parental origin of extra chromosome was assigned to 77% of detected cases. Conclusion: The PCR-based DNA diagnostic method was found to be sensitive, reproducible, and efficient, not only for diagnosis of trisomy 21, but also for tracing allelic transmission from parents to the offspring.
Introduction
T
Materials and Methods
Patients with DS (confirmed trisomy 21 cases) and their parents were enrolled in this study with prior informed consent and after approval of institutional ethics committee. Fifteen families labeled DS1-DS15, each with a confirmed DS proband, and 10 control families consisting of normal individuals were included in this study. Two milliliters of blood was collected in ethylenediaminetetraacetic acid vials. Genomic DNA was extracted using standard phenol-chloroform method with minor modifications. Samples from each patient underwent karyotyping.
Trisomy 21 detection
For molecular detection of DS two tetranucleotide microsatellite markers, D21S2055 situated at 21q22.2 and D21S11 situated at 21q21.1, were employed. For each family, both parents and the child were analyzed for both the STR markers. The primer sequences used for the amplification of marker D21S2055 were as follows: forward primer, 5′-AACAGAACCAATAGGCTATCTATC-3′; reverse primer, 5′-TACAGTAAATCAC TTGGTAGGAGA-3′. The PCR conditions for this primer set were as follows: after the initial denaturation at 94°C for 3 min, 30 cycles of PCR amplification were done (94°C for 30 s, 56°C for 30 s, and 72°C for 1 min) and final extension for 3 min at 72°C. The primer sequences used for the amplification of marker D21S11 were as follows: forward primer, 5′-GTGAGTCAATTCCCCAAG-3′, reverse primer, 5′-GTTGTATTAGTCAATGTTCTCC-3′. The PCR conditions for this primer set were as follows: after the initial denaturation at 95°C for 5 min, 30 cycles of PCR amplification were done (95°C for 30 s, 60°C for 30 s, and 72°C for 30 s) and final extension for 9 min at 72°C. PCR products were checked for amplification on 2% agarose gel electrophoresis and after staining with ethidium bromide. The amplified PCR products were resolved on a 15% polyacrylamide gel.
Detection of origin of nondisjoined chromosome
The parental origin of supernumerary chromosome 21 (therefore, the parental origin of nondisjunction) was determined after visually comparing the bands of the father, mother, and proband (Antonarakis et al., 1991; Antonarakis et al., 1993).
Quantitative fluorescent-PCR
Quantitative fluorescent (QF)-PCR refers to the amplification of chromosome-specific polymorphic microsatellite markers by using fluorescence-labeled primers, followed by quantitative analysis of the products on a genetic analyzer to determine copy number of specific chromosomal material. This approach has proven to be very useful in clinical settings, since it allows the detection of major numerical disorders in a few hours after sampling with 100% sensitivity.
We verified our results with QF-PCR results. A multiplex QF-PCR assay was applied, using Aneufast™ QF-PCR Kit. This kit contains six multiplex marker sets of STRs that can be used for amplification of selected microsatellites and the amelogenin-SRY. This combination of markers allows detection of aneuploidies involving chromosomes X, Y, 21, 18, and 13 with 100% sensitivity and specificity for nonmosaic trisomies.
Results
Cytogenetic studies
Analysis of chromosome and karyotype revealed aneuploidy of 2n = 47, +21 in probands of all 15 families. All the parents of the proband showed normal chromosome count and normal karyotype.
STR analysis
Out of 15 cases studied, we were able to detect trisomy in 13 probands through D21S11 marker or through D21S2055 or with both. The results of STR analysis are summarized in Table 1. Three distinct bands on a 15% polyacrylamide gel, except two cases in which the ratio of band intensity was 1:2, represented all detected trisomic cases. Figure 1a and b shows gel pictures in which trisomy is represented by three alleles and two alleles (1:2 ratio), respectively.
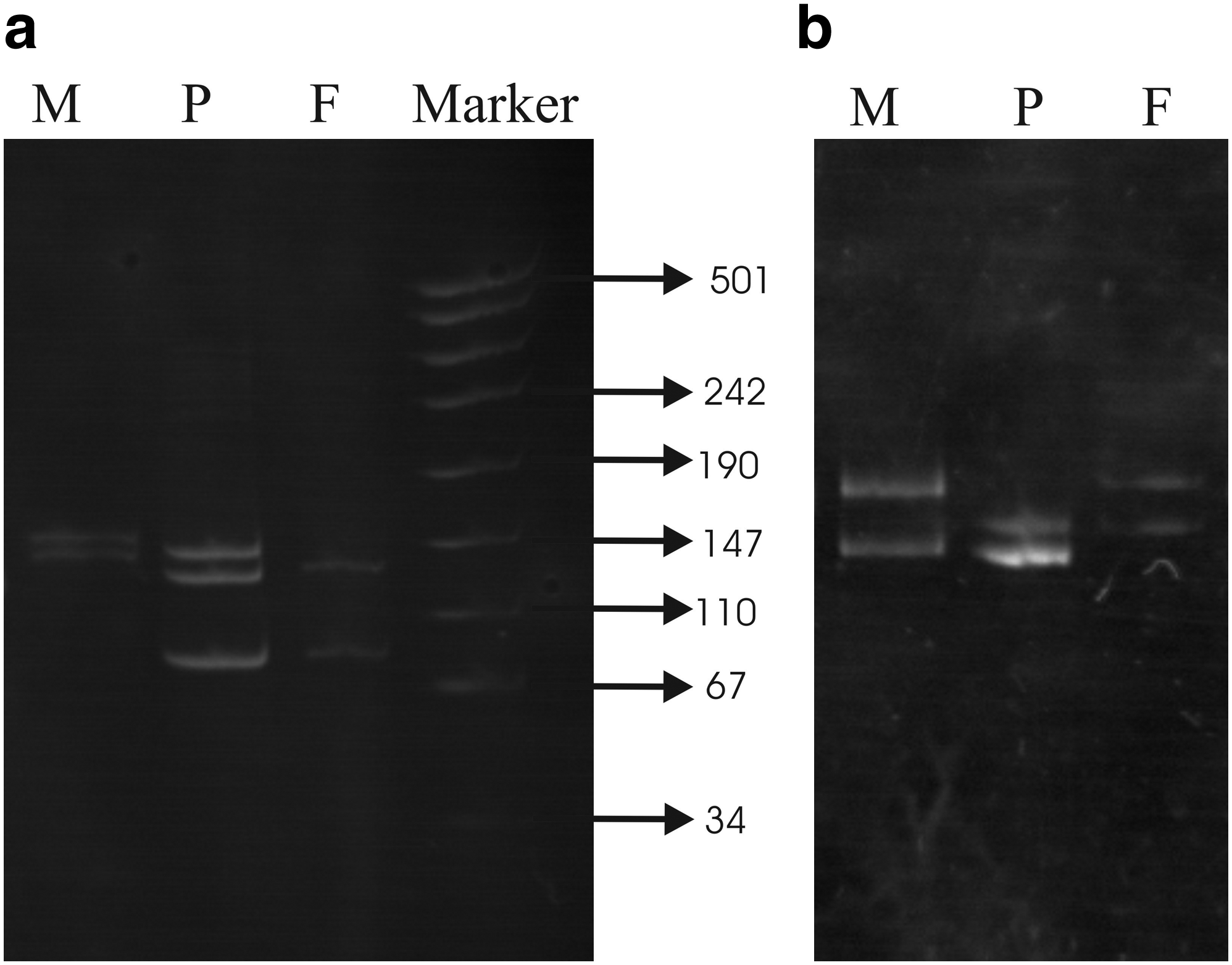
(
DS, Down syndrome.
Molecular studies of parental origin
The parental origin of the extra chromosome was assigned after the polymorphic alleles in a given family were scored. Out of 15 families analyzed we detected trisomy in 13 cases, and parental origin of the extra chromosome was assigned to all but 3 detected cases. In 8 patients the extra chromosome originated from the mother, and in 2 cases the extra chromosome originated from the father. Thus, the analysis using the microsatellite markers D21S11 and D21S2055 showed that the origin of an extra chromosome was maternal in about 80% of the cases of free trisomy 21 and paternal in about 20%.
All the alleles were scored visually and were within the range of 80-208 base pairs.
The results obtained from the above PCR method were retested using QF-PCR and results were found to be correct for all cases.
Discussion
Performing a chromosomal analysis in which culture, harvesting, slide making, banding, and reporting are intermediate steps makes laboratory diagnosis of DS complex. This procedure requires considerable time and labor. Certain indications such as prenatal diagnosis of DS require rapid diagnosis to reduce parental anxiety and since medical termination of pregnancy after antenatal diagnosis of DS is not allowed in India after 20 weeks. Rapid diagnosis becomes essential especially when the couple comes late for an antenatal diagnosis. The other method available is fluorescent in situ hybridization using uncultured cells, but this too needs proper setup and skilled personnel.
Rapid diagnosis by PCR-based methods using polymorphic STR markers may reduce these difficulties. Using this method, we were able to detect trisomy in 86.67% cases with only two markers. However, for confirmation of two trisomic cases, densitometry was needed as visual interpretation is not sufficient for diagnosis. Parental origin of the nondisjoined chromosome was assigned to 77% of the detected cases, where the ratio of maternal to paternal nondisjunction is 4:1. The reason for the two cases, which were not detected by any of the marker, may be the uninformativeness of these markers for those very loci. Clearly, the judicious choice of a few highly polymorphic markers is very essential for trisomy detection and investigation of the parental origin of trisomy 21 (Chakravarti, 1989). This method was found to be comparable to the quantitative fluorescence technique where fluorescently labeled primer, DNA sequencer, and Genescan software are usually required for genotyping (Findlay et al., 1998a; Findlay et al., 1998b; Blake et al., 1999; Valero et al., 1999). In the present study, a simple PCR-based method for trisomy detection was used where polymorphic allelic fragments were separated in polyacrylamide gel, and results were comparable to QF-PCR. Thus, this PCR-based technique can be applied for early prenatal diagnosis in resource-limited settings. Using more number of markers can further increase the reliability of the test.
Footnotes
Acknowledgments
The authors thank the families participated in the study and the professionals who helped us in collecting family history and blood sample. The project was funded (to S.A.) by Department of Biotechnology, New Delhi, India. S.J. is a Junior Research Fellow in a Department of Biotechnology project.
Disclosure Statement
No competing financial interests exist.