
Abstract
Mutations in the GJC3 gene are known to cause nonsyndromic hearing impairment (NSHI). In this study, we screened for mutations in the connexin 29 (Cx29) gene in peripheral blood collected from patients with NSHI. DNA was extracted from peripheral blood cells of 123 NSHI patients and 127 normal-hearing control subjects. Coding regions of Cx29 were amplified by polymerase chain reaction using primer pairs flanking both exons. Sequences were analyzed and compared with the published Cx29 sequence. On comparison with control subjects, only one patient and her normal-hearing mother showed a novel heterozygous variant in exon 1 c.569T>A (p. Ile190Asn), which most likely represents a rare polymorphism. From the study, we conclude that mutations in the Cx29 gene do not play a role in the causation of NSHI in Indian population.
Introduction
C
Recently, mutations in GJC3 (codes for Cx29) localized on chromosome 7q22.1 were shown to cause nonsyndromic hearing loss in a Taiwanese population (Hong et al., 2009). To date, six different mutations have been described in the GJC3 gene, which are located in the coding region of exon 1 (Yang et al., 2007; Wang et al., 2010). Cx29 is a small gene comprising two exons with an open reading frame of 840 bp. The GJC3 protein is 279 amino acids long with a molecular weight of 31.29 kDa (Sohl et al., 2001). It is expressed in cochlear Schwann cells, and required for the normal development and function of the auditory nerve (Ahmad et al., 2003; Tang et al., 2006).
In the present study, we have screened the Cx29 gene mutations in an Indian population using direct DNA sequencing. We found only one patient showing GJC3 variant among 123 patients and 127 control subjects analyzed. This is the first report to our knowledge showing the heterozygote variant c.569T>A of the GJC3 gene inherited in an NSHI family.
Materials and Methods
Nonsyndromic hearing loss was diagnosed in 123 unrelated individuals in the ENT department at Capital Hospital, Bhubaneswar. For the control group, 127 normal-hearing individuals were selected randomly. Peripheral blood was obtained with informed consent from the patients and control subjects. The genomic DNA was extracted by salting-out procedure described by Lahiri and Nurnberger (1991). The quality and quantity of purified DNA samples was determined by spectrophotometry. Primer and polymerase chain reaction (PCR) conditions for amplification of exon 1 and exon 2 covering the entire coding sequence of the GJC3 gene were selected according to procedures optimized previously (Wang et al., 2010). Bidirectional sequencing was performed for all the reactions at The Centre for Genomic Application, New Delhi. The obtained sequences were compared with the published Cx29 gene sequence (NM_181538.1). The identified variant was validated by (1) PCR-restriction fragment length polymorphism method by Tsp509I digestion of a 1008 bp amplified DNA fragment and (2) single-strand conformation polymorphism analysis of a denatured 158 bp amplified DNA fragment. Primers used for the amplification of the 158 bp DNA fragment were Cx29SSF: CCA GCT CCT TTG CAT GTC GC, and Cx29SSR: CCC CAA ACC CAG AAG CAC AAG. Approval for this study was obtained from the Institutional Human Ethics Committee.
Results
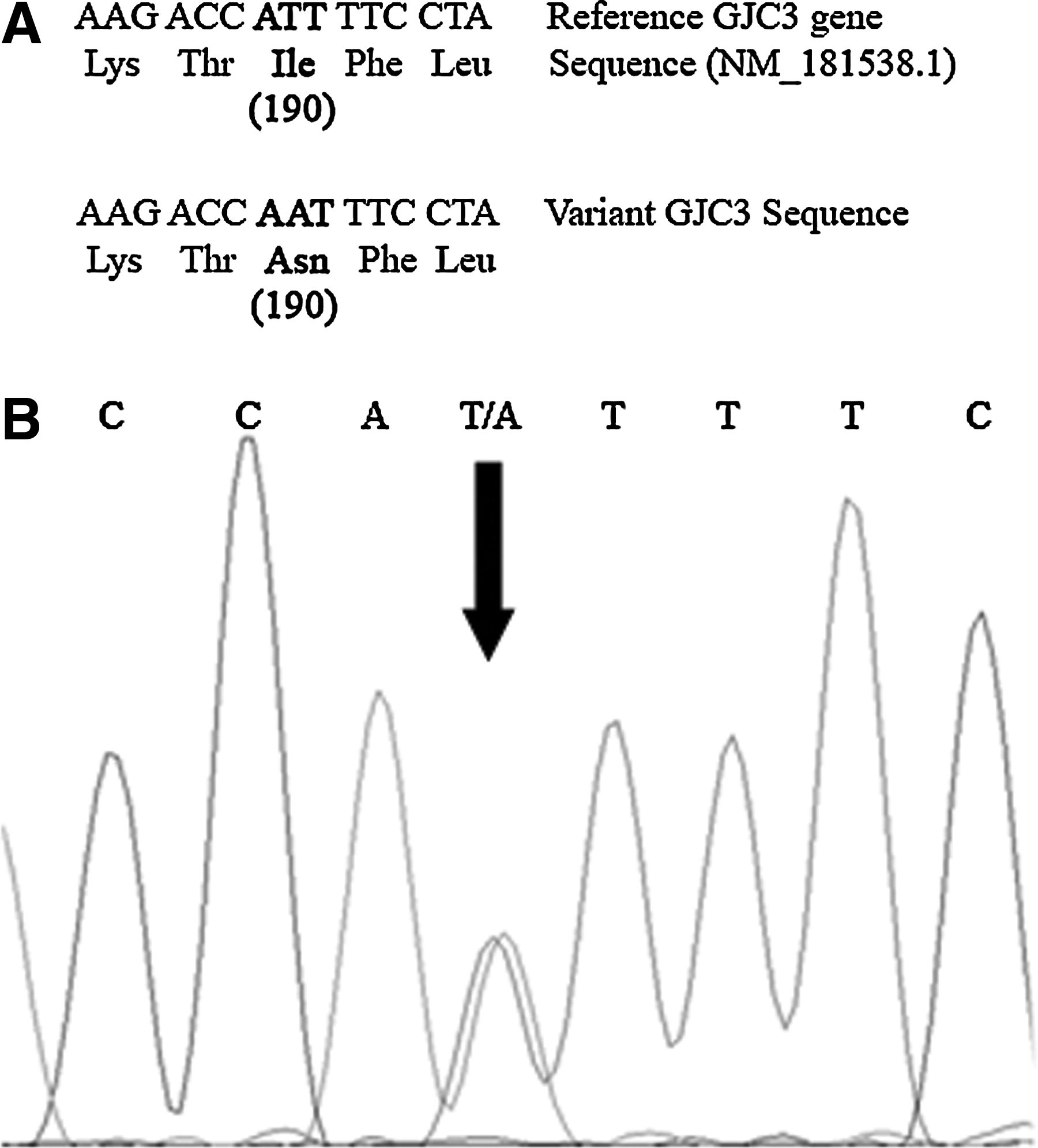
We have investigated a full spectrum of the GJC3 gene mutations in an Indian population by direct sequencing. All the samples (123 patients and 127 control subjects) screened showed the absence of mutations when compared with the reference GJC3 gene sequence (NM_181538.1). However, a heterozygous variant in exon 1 c.569 T>A was identified in a single patient. When her parents' blood samples were screened for mutations, her mother showed the heterozygous variant identical to the patient. This finding was confirmed by PCR-restriction fragment length polymorphism (Fig. 1A) and single-strand conformation polymorphism (Fig. 1B). Figure 2A shows a comparison of the reference gene sequence with the corresponding sequence in our samples. Figure 2B shows a representative sequencing data around the codon coding for amino acid 190 of Cx29.
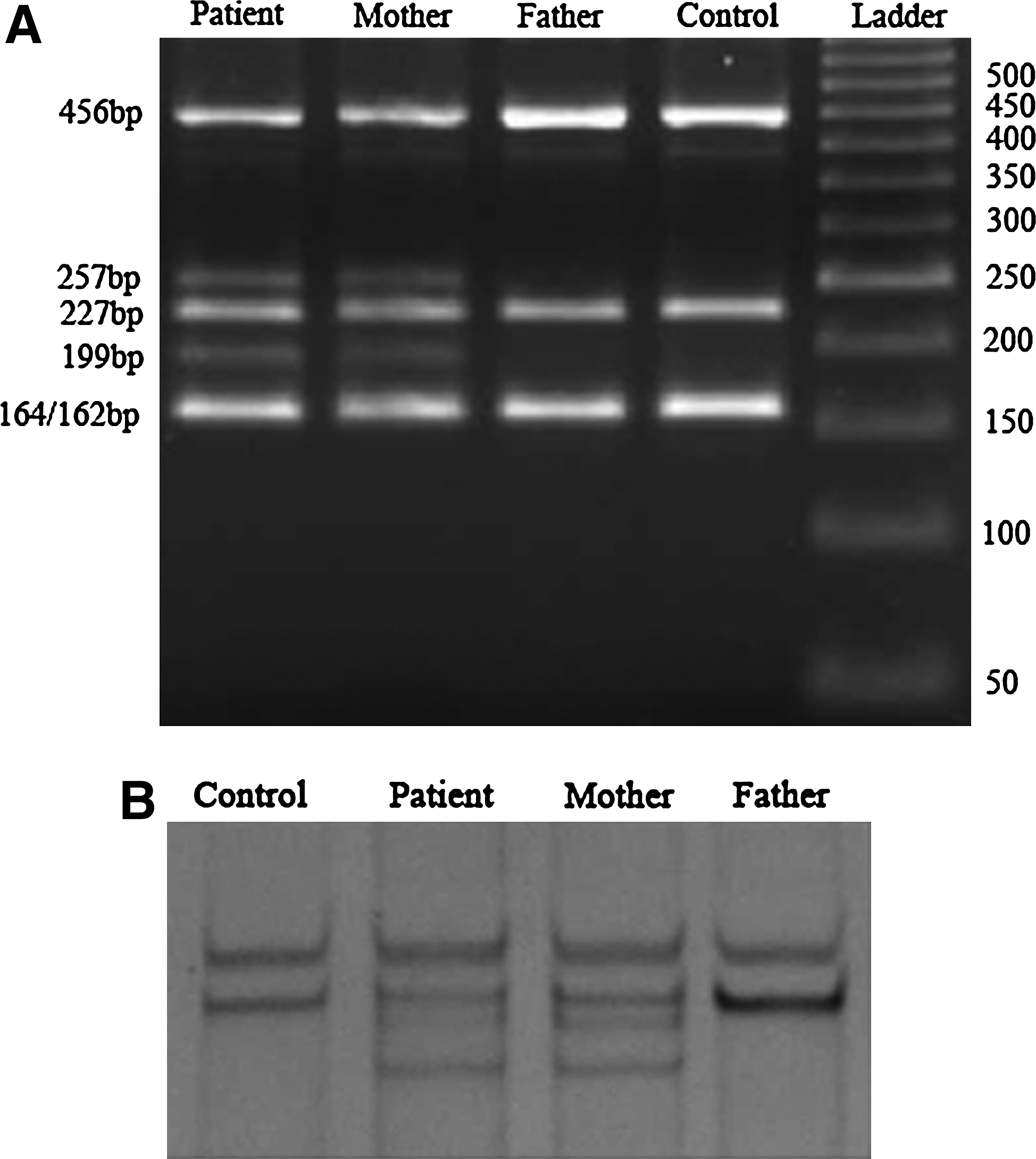
Genotyping GJC3 mutation in the patient and her family members. (
(
Discussion
It is known that mutations in Cxs (e.g., Cxs 26, 30, 31, and 43) are associated with hereditary NSHI in many of the ethnic populations studied (Hoang et al., 2009). Another member of the Cx family, Cx29, expressed in the cochlea has been reported to play a role in the development of hearing process (Ahmad et al., 2003; Yang et al., 2005; Tang et al., 2006). To investigate the role of coding mutations in the GJC3 gene in NSHI cases in an Indian population, we sequenced the entire coding region. None of the case and control samples analyzed showed any of the synonymous or nonsynonymous mutations. However, a novel heterozygote variant c.569T>A (p.Ile190Asn) was identified in exon 1 of Cx29 in a patient with nonsyndromic hearing loss. We could not detect any heterozygote variant allele among the control subjects, indicating that the polymorphism is uncommon. Yang et al. (2007) and Wang et al. (2010) reported six heterozygous mutations and two heterozygous polymorphisms in the exon 1 of Cx29 in patients with nonsyndromic hearing loss in Taiwan, but no mutation in exon 2. We failed to detect any of these known mutations or polymorphisms in our study.
Further, we genotyped c.569T>A in the unaffected parents and identified the variant in a single allele of the mother, but not the father (Fig. 1). This observation indicates that this heterozygous variant may not be associated with deafness, as it was found to be transmitting in both the affected and unaffected members. The other possibility is that c.569T>A innocuous substitution does not contribute to disease but rather represents a rare variant in the general population. The heterozygous variant c.569T>A may contribute to disease when accompanied by other heterozygous variants from the same or other genes responsible for hearing loss like the compound heterozygote c.[43C>G (+) 230G>C] reported in a Taiwanese population by Wang et al. (2010). Likewise, it is possible that this patient may have some unknown interaction with recessive alleles of other genes responsible for the disease. The presence of c.569T>A in an unaffected parent may be due to incomplete penetrance of this variant. Alternatively, c.569T>A may interact with another susceptibility variant arising de novo in the patient or inherited from the non-c.569T>A transmitting parent. Such a mechanism is supported by several genetic studies in human disorders in which double-heterozygous mutations enhance the phenotype like bone growth disorders, deafness, and hypertrophic cardiomyopathy (Flynn and Pauli, 2003; Seeman et al., 2005; van Rijsingen et al., 2009).
Using the Mutation Taster program (http://neurocore.charite.de/MutationTaster/comparison.html), we found that the alteration p.Asn190 of Cx29 is not conserved, and predicted as a polymorphism. To further investigate the effect of this variant at the protein level, both the protein structures of wild-type and mutated Cx29 were predicted by using homology modeling method. Molecular surface area (MSA) and solvent accessible surface area (ASA) were calculated for wild-type and mutated Cx29 using surface racer program (http://apps.phar.umich.edu/tsodikovlab/index_files/Page756.htm). We observed that there is a decrease in MSA and ASA due to amino acid change from isoleucine to asparagine at position 190. However, the decrease in the MSA and ASA values could be related to the function of a protein that is yet to be ascertained.
In conclusion, this study suggests that p.Ile190Asn is not a pathogenic variant and that mutations in the Cx29 gene do not play a role in the causation of NSHI in the Indian population.
Footnotes
Acknowledgments
We thank all the subjects who participated in the present study, and Pragyna Priyadarshini for technical assistance. This work was supported by the Department of Science & Technology (DST), New Delhi, Government of India (Grant Sanction No. SR/SO/HS-61/2007 dated 23.10.2007).
Disclosure Statement
No competing financial interests exist.