
Abstract
In β-thalassemia, point mutations in the β-globin gene are largely responsible for either decreased or no β-globin synthesis. The β-globin gene has three exons and two introns. The molecular characterization of β-thalassemia is absolutely necessary for carrier screening, for genetic counseling, and to offer prenatal diagnosis. The objective of the present study was to identify the rare mutations in β-globin gene of β-thalassemia patients. We have sequenced the entire β-globin gene in 36 clinically identified thalassemia patients from the Karnataka region using polymerase chain reaction and sequencing. Our analysis revealed 11 β-thalassemia variants. The most common being IVSII-16 G>C, IVSI-5G>C, IVSII-74 T>G, codon 3 (T>C), and Poly A site (T>C). In addition, we have also documented a novel deletion at codon 6 (-CT) (HBB:c.16delCT). These data are useful in future molecular screening of the population for implementing a thalassemia prevention and control program. Further it is found that family studies and comprehensive hematological analyses would provide useful insights for accurate molecular diagnosis of thalassemia phenotype and offers an interesting subject for further investigations in the Indian populations.
Introduction
β-
The Indian subcontinent is inhabited by nearly 5000 anthropologically well-defined indigenous populations, which are from diverse ethnic, genetic, tribal, cultural, and social background (Singh, 2003). The caste endogamy and clan exogamy, which are practiced among different communities since time immemorial, might have led to a conservation of gene pools among them (Bhasin et al., 1994). This in turn might have resulted in accumulation of mutations leading to increased frequency of genetic disorders (Kivisild et al., 2003). The initial studies in the Indian population on β-thalassemia were undertaken in the individuals from the states of Gujarat and Punjab and the Sindhi community. Many of these groups who originated in Pakistan and are also living in other countries revealed five major mutations, IVSI-5(G-C), IVSI-1(G-T), 619-bp del, codon 41/42 (-TCTT), and Codon 8/9 (+G), which accounted for 90% of all mutations (Kazazian et al., 1984; Thein et al., 1988). Later studies that focused on the populations of Gujarat, Punjab, and Maharashtra are consistent with the results of the previous studies (Varawalla et al., 1992; Garewal et al., 1994). Further studies revealed wide distribution and extensive heterogeneity of β-thalassemia mutations in different subpopulations of India (Gupta et al., 2003; Bashyam et al., 2004; Sinha et al., 2009a, 2009b; Agarwal et al., 2010; Kumar and Agarwal, 2011) and found that the highest percentage of rare mutations was from the Karnataka region (Colah et al., 2009).
A large number of studies have been carried out in North Indian populations (Gupta et al., 2003; Sinha et al., 2009a, 2009b; Agarwal et al., 2010; Kumar and Agarwal, 2011). The Indian population is comprised of different ethnic groups with much genetic heterogeneity (Reich et al., 2009). The population of North India is largely Indo-European and hence different from other regions of the country. Therefore, previous studies may not be sufficient to describe the complete spectrum of HBB gene mutations in this country. Very little data about HBB gene mutations in the population of South India are available so far. Therefore, the purpose of this study was to determine the spectrum of β-thalassemia mutation in the population of the South Indian region.
Subjects and Methods
Thirty-six thalassemia patients from the Karnataka region, who regularly attend the Karnataka Institute of Medical Science, Hubli and JJM Medical College, Davanagere, during January 2008 to February 2010, were included in the present study. All the thalassemia patients were confirmed with hemoglobin electrophoresis (Interlab Genio S Electrophoresis, Via Rina Monti, Roma, Italy). All patients were also transfusion dependent. Blood samples (5.0 mL) from each individual were collected just before the transfusion, with the informed written consent of all patients who are above 18 years or parents of individuals who are <18 years old following the institutional ethical guidelines. Genomic DNA was extracted from all participants using a standard procedure described elsewhere (Thangaraj et al., 2002).
Sequencing of HBB gene
The complete nucleotide sequence of the gene encoding β-globin was amplified using six overlapping primers (approximately at every 300-350 bp interval) that were designed using Primer 3 software (based on sequence NM_000518). The sequence of the primers will be available on request from the authors. Amplifications were performed in a 10 μL volume containing one unit of AmpliTaq Gold (PerkinElmer, Wellesley, MA), 1×polymerase chain reaction (PCR) buffer, 1.5 mM MgCl2, 200 μM deoxynucleotide triphosphates, 1 pmol each of forward and reverse primer, and 40 ng of genomic DNA. PCR products were visually verified on 2% agarose gels and directly sequenced using a BigDye Terminator cycle sequencing kit and an ABI PRISM 3730 DNA analyzer (Applied Biosystems, Foster City, CA).
Results
Ten mutations found in the patients are shown in Table 1. Among 36 samples analyzed, 13 showed IVSII-16 G>C, 10 of IVSI-5G>C, 8 of IVSII-74 T>G, 7 of codon 3 (T>C), 3 of Poly A site (T>C), and 1 each of codon 6 (-CT), codon 16 (G>A), codon 31 (G>C), codon 31 (G>A), and IVSII-837 T>G. In the present study, we documented a novel frameshift deletion (-CT) at codon 6 in one sample [Codon 6 (-CT)] (Fig. 1). IVSII-16 G>C is the predominant mutation (36%) followed by IVSI-5G>C (28%), IVSII-74 T>G (22%), codon 3 (T>C) (19%), and Poly A site (T>C) (8%).
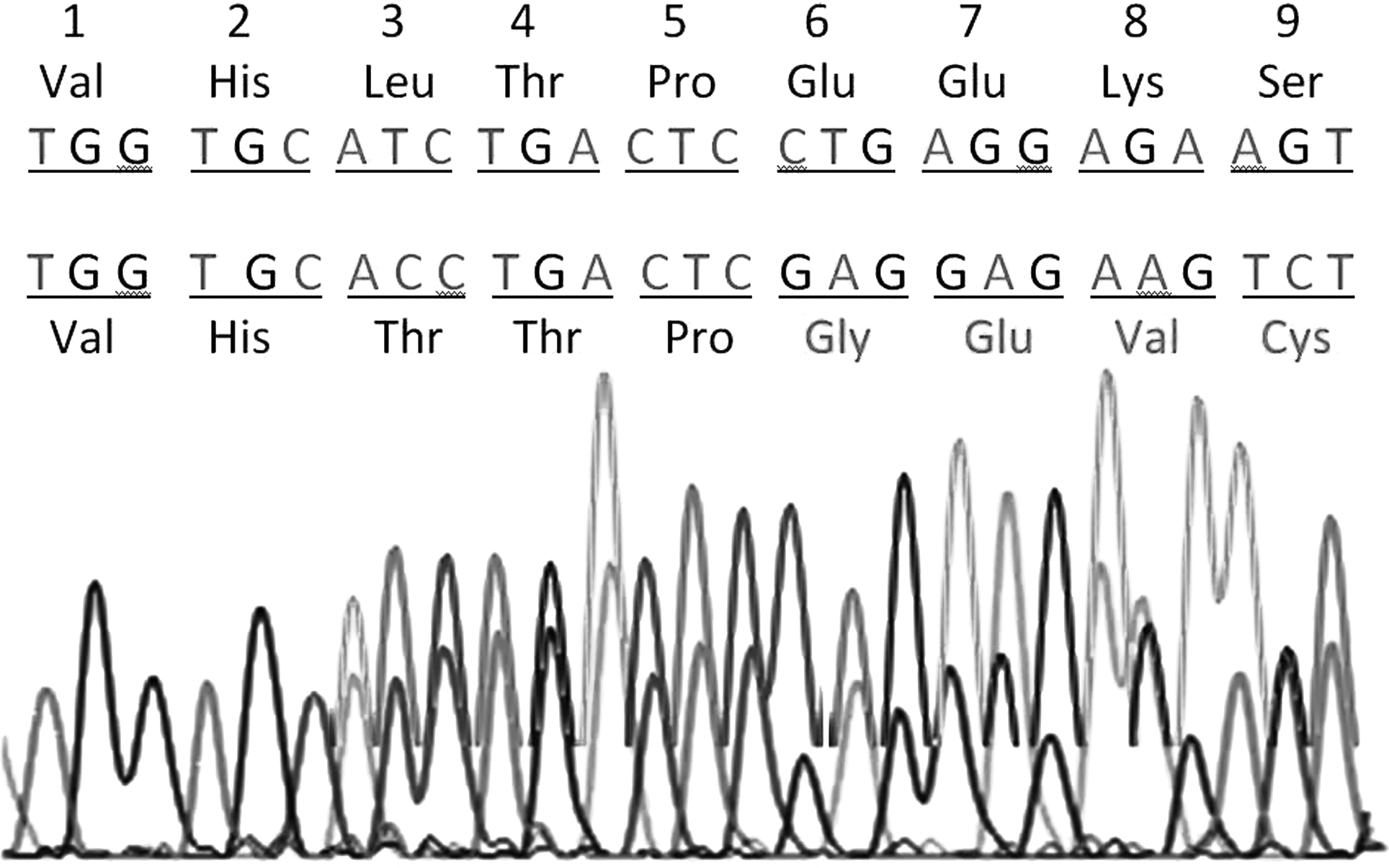
Nucleotide sequence of the proband β-globin gene, codons 1-9. The upper sequence is normal. The lower sequence shows a mutation at codon 3 (ATC>ACC) and novel frameshift change, starting at codon 6 deletion of CT in codon 6 (CTG>GAG).
HGVS, Human Genome Variation Society.
Discussion
Although several causative mutations have been reported to be associated with β-thalassemia syndromes, the spectrum of mutations and their frequencies in most populations usually consist of a limited number of common mutations and a slightly larger number of rare mutations (Hardison et al., 2002). Sequence analysis of the HBB gene in the populations of the Karnataka region revealed that among some common mutations, IVSII-16 G>C is the predominant mutation (36%), followed by IVSI-5G>C (28%), IVSII-74 T>G (22%), codon 3 (T>C) (19%), and Poly A site (T>C) (8%) and a few rare mutations: IVSII-16 G>C, IVSI-5G>C, IVSII-74 T>G, codon 3 (T>C), Poly A site (T>C), codon 6 (-C), codon 6 (-T), codon 16 (G>A), codon 31 (G>C), IVSII-837 T>G, and codon 31 (G>A) (3% each). Interestingly, for the first time we have found two deletions at codons 6 (-C) (c.16delC) and 6 (-T) (c.18delT) in one patient whose beta thalassemia was diagnosed in 1998 when she was 4 months old and till now she is dependent on blood transfusion. This girl presented with peculiar and hematological features such as progressive pallor, frontal and parietal bone prominence, depressed nasal bridge, prominent malar eminence, maloccluded teeth, hyperpigmentation of palm, sole, and skin due to hemochromatosis, hepatomegaly, and stunted growth and hemoglobin electrophoresis shows HbA 95.1% and HbA2 4.9%.
IVSII-16 G>C is in the alternative splicing region of mRNA, which might lead to differential expression. Chinese IVSII-16 C>G subjects have shown a greater improvement of running economy indexes after 18-week 5000-m running program than those with other genotypes (He et al., 2006). This polymorphism is quite common in Indian populations. Fifty-five populations that were studied all over India have revealed this C allele frequency to be ranging from 0.21 in Indo-Europeans to 0.66 in Dravidian linguistic groups. IVSI-5(G>C) was found in an Asian Indian (Kazazian et al., 1984) and in a Chinese (Cheng et al., 1984). Although this is the second major mutation in the present study, this accounts for 54.7% of all β-thalassemia alleles nationally, and the majority of subjects with this mutation originate from or are residents of the major states of Maharashtra, Gujarat, Uttar Pradesh, and West Bengal (Agarwal et al, 1997, 2000; Kukreti et al., 2002). IVSI-5 (G>C) frequency varied from 15% in the immigrant population from Pakistan and Goa on the west coast to 88.6% in the eastern state of Orissa (Colah et al., 2009). Codon 3 (T>C) mutation changes the third codon of the HBB gene from CAT to CAC (rs713040), both coding for histidine. This silent mutation with no apparent effect in thalassemia has been observed in patients of Bangladeshi origin (Ibn Ayub et al., 2010).
The Poly A site mutation, AATAAA→AACAAA, was reported earlier in American Blacks (Gonzalez-Redondo et al., 1991). Poly A site (T>C) is a rare mutation in Indian populations (Gorakshakar et al., 2004). The rare codon 31 (G>C) (c.92G>C) mutation found in only one patient completely prevented mRNA splicing and could not produce mRNA in carriers of this mutation as previously reported in a Turkish thalassemic patient (Tadmouri et al., 2000) and a southern Iranian patient (Rahimi et al., 2006). Another rare codon 31 (G>A) (c.92G>A) mutation found in only one patient has been previously reported in a southern Iranian patient (Karimi et al., 2002). Codon 15 (G>A) (c.47 G>A) mutation TGG>TAG, a change of one nucleotide, results in the termination of translation (Gray et al., 1995). The novel codon 6 (-CT) frameshift mutation nucleotide sequence was translated to amino acids and compared with the reference sequence. There is a mismatch of 57% in these two sequences when compared globally in EMBOSS pairwise alignment tool. This frameshift mutation leads to abnormal splicing of β-globin mRNA, resulting in the formation of a nonfunctional β-globin gene product (Fig. 1). The present study reports a novel mutation; the remainder is atypical in Indians. This indicates the increasing collection of molecular defects causing β-thalassemia in Indian populations. According to previous reports, 11 HBB mutations have been shown to be common in India. Among these 11, 6 account for about 94% of all positive cases. These include IVS1+1G>T, IVS1+5G>T, c.124_127delTTCT, c.47G>A, c.366_494del, and c.20A>T. The other two less common mutations are c.27_28insT and c.79G>A.
The spectrum of mutations that are reported in this study confirm the heterogeneity observed in the different population groups within India (Bhasin et al., 1994; Edison et al., 2008; Colah et al., 2009; Sinha et al., 2009b). Population screening associated with genetic counseling is particularly helpful by allowing families at risk to make informed decision on their reproductive choices.
Footnotes
Acknowledgment
This research was supported by the Department of Medical Education and Department of Higher Education, Karnataka Government, Karnataka, India.
Disclosure Statement
No competing financial interests exist.