
Abstract
Current studies of human genetic diversity are focused in two areas: first, detection of rare mutations in highly selected clinical cases; and second, in common single-nucleotide polymorphism (SNP) and haplotype effects in the general population. Less frequent SNPs and “paucimorphisms” remain underexplored, although lower frequency coding SNPs are more likely to have functional impact. We have developed a cost-efficient mutation scanning technology, meltMADGE, for population mutation scanning. Previous research in GHR has explored its role in extreme (−3 SD) growth retardation and, subsequently, “moderate” (−2 SD) growth retardation cases. Here, we describe meltMADGE assays for the entire coding region of GHR. As a first step we have established long polymerase chain reaction subbanks for GHR from 2423 unselected subjects and have applied meltMADGE scanning assays of exons 4 and 5 to these subbanks. A novel paucimorphism present at 439+30A>C (allele frequency: 0.0021) in intron 5 (location chr5:42,695,221 in GRCh37/hg19) was identified in 10 individuals, confirmed by sequencing and analysis made for major phenotypic effects. This approach is relevant to the deep sampling of populations for less frequent sequence diversity, some of which is expected to exert significant phenotypic effects.
Introduction
S
GHR is located on chromosome 5p (Barton et al., 1989), spans 85 kp, and includes nine coding exons. Exon 2 encodes an 18-amino-acid signal sequence, exons 3-7 encode the extracellular domain, exon 8 encodes the transmembrane domain, and exons 9 and 10 encode the intracellular domain (Meacham et al., 1993), including a proline-rich region.
Over 50 mutations have been reported (Milward et al., 2004). These mutations include large and small deletions, splice site mutations, and nonsense and missense single-base codon mutations. Earlier literature focused on classical Laron syndrome inherited in an autosomal recessive fashion. In general, this spectrum of mutations (Amselem et al., 1993) results in absence of receptor function. Haploinsufficiency in heterozygotes for such mutations does not produce a clinically recognizable phenotype. Studies of heterozygous Ecuadorians for splice mutation E180, which has been shown to be functionally null, did not identify any phenotypic effects in heterozygotes (Rosenbloom et al., 1998). However, expression of a defective receptor may exert dominant negative effects (Ayling et al., 1997; Dos et al., 2004). The growth hormone receptor is expressed on the cell surface and receptor molecules dimerize in the process of binding GH and inducing signal transduction. Defective receptor molecules can thus interfere with wild-type molecules when defective and wild-type molecules dimerize. Additionally, if defective molecules have long half-lives at the cell surface, this will further contribute to dominant negative effects. A mutation (871-1G>C) leading to exon 9 splice skipping exemplifies these effects (Ayling et al., 1997). Further, presence of variant forms of or differential release of the extracellular domain of the growth hormone receptor into the circulation might also have quantitative effects. A common polymorphism involves the presence or absence of (all of) GHR exon 3, which encodes part of the extracellular domain of the receptor. Compared with wild type, heterozygotes show a different responsiveness to exogenous growth hormone, representing another example of the “dominant” or “quantitative” effects that expressed GHR can exert. Most mutation studies have been undertaken in extremely growth-retarded children (>3 SD below the mean), although one study demonstrated the occurrence of amino acid mutation in children selected for moderate (−2 SD) growth retardation (Goddard et al., 1995). However, no studies of the full spectrum of sequence variation, which may be present in the general population, or studies of their potential quantitative effects have ever been conducted.
In this article, we describe development of mutation scanning assays of GHR using a system that we developed to be capable of high throughput and economy, which are necessary for large-scale population-based mutation scanning. We present scans for exon 4 and exon 5, representing a functionally important part of the receptor, in a population sample of 2577 anthropometrically characterized subjects.
Methods
Participants and ethics statement
The genomic DNA banks represented previously described cohorts recruited in Hertfordshire, United Kingdom. The first DNA bank represented 663 men and 449 women, 1112 subjects in total (from the North and East regions). The second bank represented a further 750 men and 690 women, 1440 subjects in total (from the East region). These cohorts and their clinical characterization have been previously described (Syddall et al., 2005 and references therein). Ethical approval was from the Hertfordshire and Bedfordshire Local Research Ethics Committee (reference EC98248) for the second bank and from East and North Hertfordshire Health Authority for the first bank (1980s). Written informed consent was obtained for the second bank (1990s-2000s); verbal informed consent was obtained for the 1980s bank, with explanation of the study in initial contact by letter, followed by interview at home of those consenting. Participants were then invited (apart from a few who were housebound or on warfarin therapy) to attend a local clinic for venesection for hematological, biochemical, and genetic studies, which most did attend. All research studies were conducted on anonymized data. Parameters relevant to the present study were anthropometric measures as older adults.
DNA bank and GHR long-range PCRs
Six pairs of long-range primers for GHR were designed using the Primer3 program (Table 1). Pairs of primers were optimized to get clear, low-background bands on agarose checking gels. Long PCR subbanks were then prepared for the regions around exons 4 and 5 by amplifying six sets of 7×384-well plates with long-range primers. Two microliters of 1/1000 dilution of long PCR product was used as template in a 10 μL melt MADGE PCR.
Amplification of GHR exons
We adopted the set of primer sequences used in DGGE analysis of GHR (Amselem et al., 1993), but GC clamps were lengthened to be consistent with our own laboratory protocols. For each exon, we also designed a pair of artificial positive control primers with a single-base mismatch near the 3′ end of the shorter primer. Each pair of primers was optimized to get clear, low-background DNA bands on MADGE PAGE nondenaturing checking gels. Reactions were then run using the relevant long PCR subbank of 7×384-well plates.
Melt-MADGE analysis
Equipment
Melt-MADGE analysis is based on a real-time variable-temperature electrophoresis system essentially as previously described (Alharbi et al., 2005). In brief, it includes a 10×10×15 cm electrophoresis tank with system for ramping the buffer temperature in real time. The tank accommodates up to 11 urea MADGE gels poured in a purpose-built box.
Optimization of melt-MADGE assay for each exon
First, the melt temperature (Tm) of each exon was calculated using an in-house program acting as a windows interface to Melt87 (Lerman and Silverstein, 1987). Then, three constant temperature electrophoreses were conducted on 5% polyacrylamide gels containing 4 M urea for each exon at the temperatures of Tm−5°C, Tm, and Tm+5°C. Finally, the velocity-temperature charts were drawn for each exon and potentially suitable temperature ramps for melt-MADGE were determined.
Artificial, positive “heterozygote” controls were generated by mixing 4 μL of wild-type PCR product and sample and 4 μL of mutated primer-PCR product (single-base mismatch to wild type). The mixtures were denatured at 95°C for 5 min, put on ice for 2 min, and then held at room temperature for 1 h.
Wild-type and artificial “heterozygote” controls were electrophoresed on a 5% polyacrylamide gel containing 4 M urea in the melt-MADGE system using the most appropriate thermal ramp. According to these results, some small refinements of temperature, concentration of polyacrylamide, urea, or electrophoresis time might be made.
Melt-MADGE scanning of GHR in population samples
For each exon, PCR products from seven 384-well subbank plates were run in a melt-MADGE analysis system. The PCR products of each 384-well plate were loaded on four 96-well urea MADGE gels along with artificial heteroduplex control samples. Band detection was performed by Vistra Green staining and Fluorimager 595 scanning.
Sequence analysis
Each sample with variant DNA patterns (more than the one wild-type band) in the melt-MADGE assay was sequenced using a standard shrimp alkaline phosphatase and exonuclease I and Bigdye Sequencing Kit (ABI) protocol. Sequencing products were purified by ethanol precipitation and were redissolved in 25 μL template compression reagent (ABI) prior to analysis on an ABI 310 automated sequencer (ABI). Sequences were aligned and compared using Lipman and Pearson's Align Program (http://molbiol.soton.ac.uk/compute/align.html).
Results
The development of melt-MADGE assays for GHR mutation scanning
For each exon of GHR, a temperature-velocity chart was drawn according to three electrophoresis results at predicted Tm+5°C, Tm, and Tm−5°C. The temperature range with rapid mobility change was thus determined. The plot for exon 5 is shown as an example (Fig. 1). The predicted Tm was 61°C for this fragment after correcting for use of the 4 M urea. Therefore, the temperatures for the three electrophoreses are 56°C, 61°C, and 66°C. An obvious mobility shift was observed (Fig. 1) between 56°C and 66°C, with gel migration velocity reduced from 7 to 3 mm/h. The result showed that this temperature range was sensitive for melting analysis of exon 5, and it was trialed as the initial temperature ramp. Using an artificially constructed “heterozygote” PCR co-annealing, it was found that a 5°C-7°C ramp was the most effective in achieving good resolution, typically from about Tm−5°C to Tm+1°C. Most artificial positives resolved to a four-band pattern (some to three if the homoduplexes did not resolve), whereas wild-type produced one band. The optimized melt-MADGE assay conditions for exons 2-9 inclusive of overlapping for three amplicons representing the coding region of exon 10 are given in Table 2. For each exon, good band patterns and resolution were obtained for both wild-type amplicons and an artificial positive “heterozygote.”
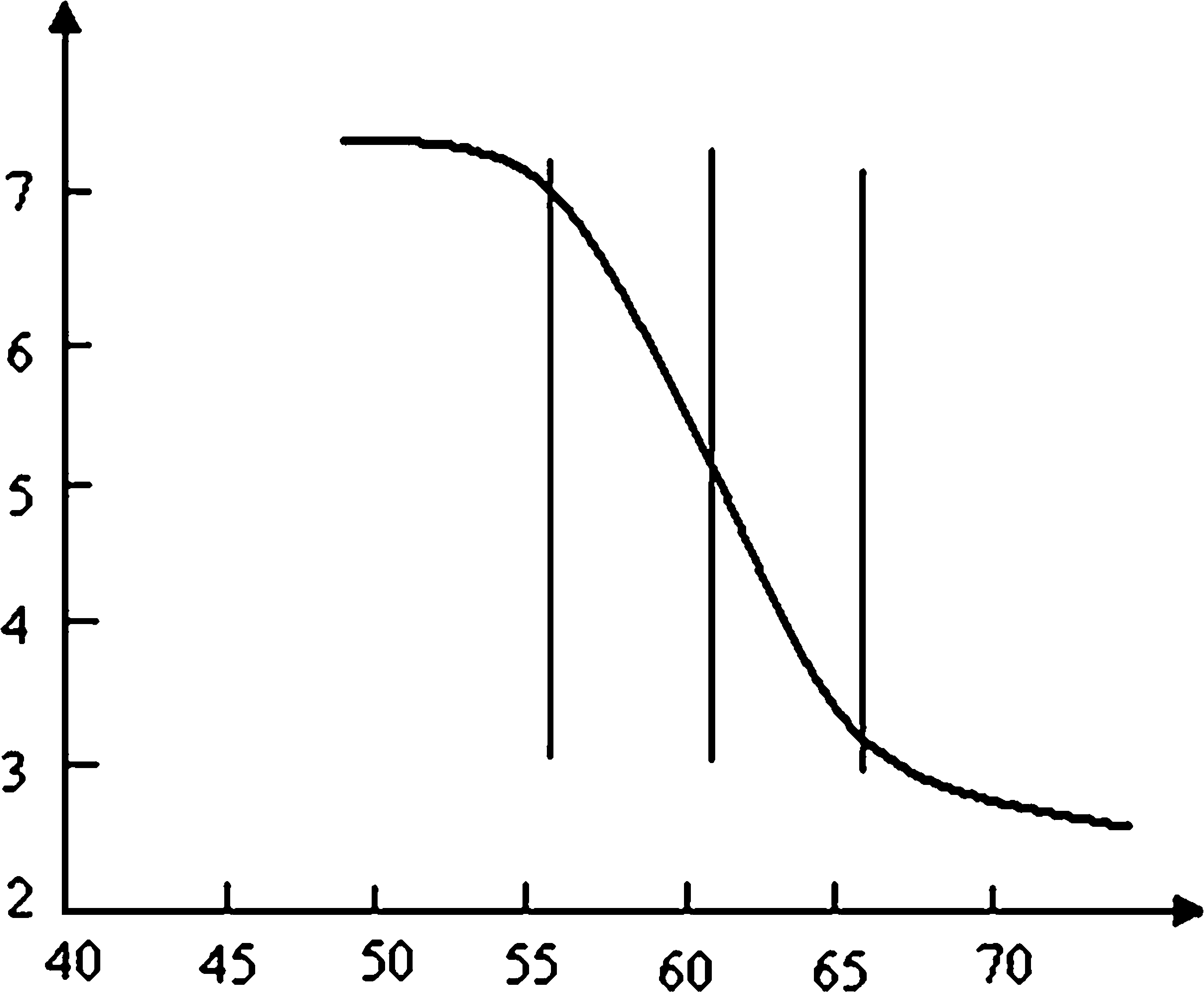
Velocity-temperature chart of exon 5 amplicon of GHR gene in melt-MADGE assay.
Including GC clamps.
Corrected for 4 M urea.
PA, polyacrylamide gel.
Variant band patterns for exon 5 amplicon on melt-MADGE gels
Ten individual subjects with the same variant pattern were identified on melt-MADGE gels. Wild-type samples presented one-band pattern (homoduplex) on the melt-MADGE gels, but these 10 DNA samples showed a four-band pattern. An example is shown in Figure 2. All 10 variant amplicons were subjected to sequence analysis.

Image of one array for GHR exon 5 amplicon melt-MADGE analysis.
Sequence analysis of the 10 DNA samples with variant band patterns
Direct sequencing showed that all 10 DNA samples possessed the same A>C base at 439+30A>C in intron 5 (location chr5:42,695,221 in GRCh37/hg19) (Fig. 3B). This paucimorphism has not been previously described and has an allele frequency of 0.0021.
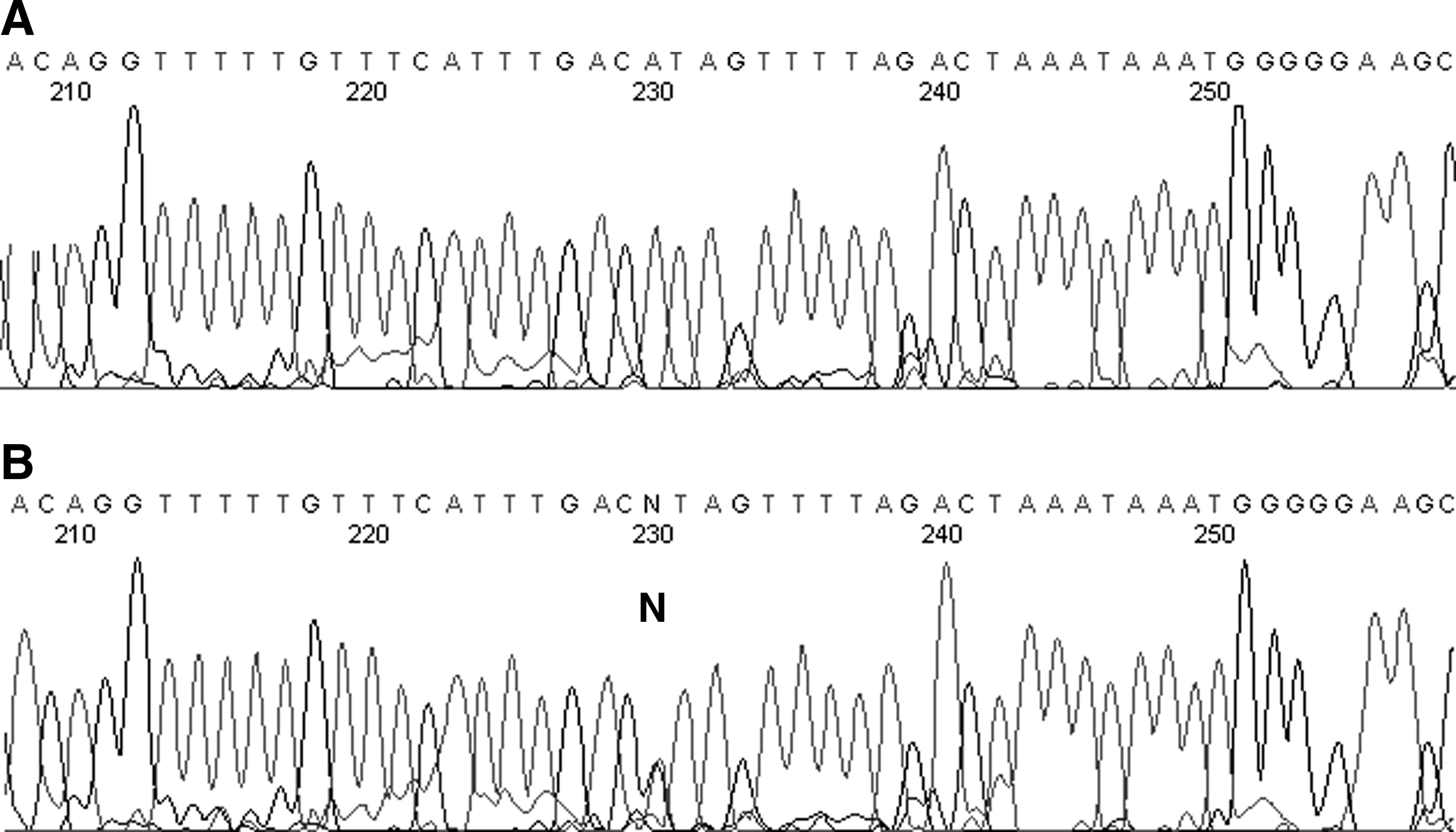
Sequencing results for one sample with wild type
Phenotypic characteristics of 439+30A>C carriers
Sex-specific Z-scores were derived for weight, height, waist, waist-hip ratio, and body mass index in relation to the relevant cohort. For height, these were −1.39, −0.78, −0.93, −0.62, −0.93, 1.96, −0.10, −0.64, −0.40, and −0.53. For weight, these were −1.06, −0.28, 1.73, −0.65, −0.82, 1.04, −0.017, −0.40, −1.09, and 0.11. Note that values between −1.96 and 1.96 represent 95% of observations, that is, these mutation carriers are not outliers for either trait, and although there was a slight bias toward negative values for height, there was no significant difference for this or the other traits (data not shown).
Discussion
We have developed melt-MADGE assays for mutation scanning of the entire GHR coding region. These assays have the characteristics of high throughput, low cost, and easy operation, which will be suitable for population mutation scanning without requirement for high capital expenditure. Scanning of amplicons spanning exons 4 and 5 (where mutations are prevalent in Laron Syndrome) identified a novel paucimorphism in 10 subjects in a population sample of 2423 subjects.
The characteristics of melt-MADGE assays have been previously examined in studies of LDLR and BRCA1 (Alharbi et al., 2005) and MC4R (Alharbi et al., 2007). In brief, most single-base mutations and small indels are detected by these assays. In contrast to denaturing HPLC or direct sequencing, overall cost is a fraction (e.g., 1/7th) of PCR cost rather than a multiplex (e.g., 30 times) of it (Alharbi et al., 2005). This lends the technique well to scoping the full range of sequence diversity through to singleton, paucimorphic, or low-frequency polymorphic alleles in the general population. Our identification here of a previously unknown GHR intron 5 paucimorphism 439+30A>C (location chr5:42,695,221 in GRCh37/hg19) in 10 of 2423 subjects (allele frequency 0.0021) exemplifies the potential.
In this development we used a systematic approach to assay optimization. In every case, an artificial (primer generated) positive control was established by co-annealing “wild-type” and “artificial” amplicons. Three constant temperature runs were then tried, Tm−5°C, Tm, and Tm+5°C. Usually, this confirmed that a ramp Tm−5°C to Tm+1°C was suitable for resolution of heteroduplexes.
For large-scale scans for rare sequence variants in finite DNA banks, DNA template consumption will be a significant problem. Possible conservative approaches include degenerate oligoprimer preamplification (Zhao et al., 1998), phi 29, or Rubicon preamplification (www.genengnews.com/gen-articles/whole-genome-amplification-technologies/854/). For focused studies of specific genes, the establishment of long PCR subbanks is an alternative. In this study, we developed five long PCR subbanks from 2423 subjects, giving complete tiling of the gene from exon 2 to exon 10. At 1/100-1/1000 dilution, these banks would support any future scanning or resequencing of exons or introns.
Scanning of populations for rare sequence variants, potentially to describe the full depth of sequence diversity and its contributions to the full range of phenotypic diversity, has so far received little attention. The 1000 Genomes project (www.1000genomes.org) will soon change this for discovery of less frequent sequence variation genome wide, but the study of phenotyped collections presents limitless potential. Selection of trait extremes for HDL-cholesterol from a population sample followed by directed sequencing of three genes relevant to HDL-C has been undertaken (Cohen et al., 2004) and exemplifies one approach to relating intermediate traits to their underlying mutational spectrum. However, the costs and time for direct resequencing even by automated multicapillary systems somewhat preclude full-population studies. This leaves scope for efficient scanning methodologies that can identify rare variants for sequencing while evading resequencings of the wild type; and until whole genome or exome sequencing is routinely affordable on a cohort-scale basis, focus on specific genes of major interest (as for SNPs identified in genome-wide association studies for complex traits) will continue to be necessary. It has been noted from a combination of theoretical and empirical data that missense alleles with a frequency of 6% or less are more likely to exert phenotypic effects than common protein polymorphisms (Wong et al., 2003). It also appears that synchronous duplex and multiplex point mutations within close syntenic range are quite common both somatically and in the germ line (Wong et al., 2003). Syntenic paucimorphisms or rare mutations can occur in complete linkage disequilibrium (Alharbi et al., 2005 and references therein). Rare alleles, therefore, merit greater focus of attention.
GHR intron 5 paucimorphism 439+30A>C occurs in a generally %AT rich and T-motif-rich region. Such regions have been shown to increase efficiency of expression in both mammals and plants, for example, in HBB in man (Bharadwaj et al., 2003) and maize (Clancy and Hannah, 2002). However, anthropometric phenotypes in these subjects were not significantly different from wild type; therefore, this particular paucimorphism appears to be silent or of unmeasurable minor effect. The mutation is in intronic sequence near the exon but not in a location predicted to influence pre-mRNA splicing. Although the assay focus was on exonic sequence and splice sites, a small amount of intronic sequence is necessarily included in the PCR amplicons. It is notable that no coding sequence variation was identified in 5154 alleles in this study. For LDLR, we previously estimated that 4/5 of all amino acid changes do not cause the severe hypercholesterolemia of familial hypercholesterolemia (Day et al., 1997). A comparable overrepresentation of codon mutations relative to stop codons appears to occur in Laron syndrome (Amselem et al., 1993). However, for any given exon, amino acid changes of moderate, no, or protective effect are only predicted to occur at a frequency of one per few thousand alleles. Very large population studies will be needed to define the “reference range” of such sequence variation, which will be valuable in the interpretation of missense mutations in case diagnostics.
In conclusion, we have demonstrated the potential of melt-MADGE assays of GHR to explore the full sequence diversity of this gene in the general population.
Footnotes
Acknowledgments
G.H. was the recipient of a Government of China Scholarship. The Hertfordshire cohort studies and original development of meltMADGE were funded by grants from the Medical Research Council of the United Kingdom.
Disclaimer
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure Statement
No competing financial interests exist.