
Abstract
Background: Hemoglobinopathies are a very heterogeneous group of congenital hemolytic anemia. Aim of the Study: Was to detect the prevalence of hemoglobinopathy disorders in adult patients sent for diagnosis of anemia by high-performance liquid chromatography techniques in Saudi Arabia.
Introduction
Individuals affected with sickle cell disease have lifelong hemolytic anemia with acute and chronic tissue damage secondary to the blockage of blood flow produced by the abnormally shaped red cells. Sickle cell disease can be lethal, especially in early infancy or childhood (10% mortality) because of overwhelming sepsis or splenic sequestration. Current predictions indicate an 85% chance that infants with homozygous sickle cell disease (HbSS) will survive to at least 20 years of age. The sickle gene is commonest in Africans (up to 25% gene frequency in some populations), but it is also found in India, (Munshi et al., 2009), the Middle East, and Southern Europe (Bardakdjian and Wajcman, 2004).
The thalassemia syndromes are characterized by absent or deficient synthesis of one or more of the normal globin chains. This contrasts with the other hemoglobinopathies in which the variant hemoglobins are qualitatively or structurally abnormal. The major causes of mortality are iron overload and overwhelming infections after splenectomy, the former due to excessive iron deposition as a result of blood transfusions and increased gastrointestinal absorption. β-thalassemia trait is generally not detected in the newborn period, but anemia becomes evident in the first 1-2 years of life. Homozygous β-thalassemia (β-thalassemia major) is a life-threatening condition that requires chronic transfusion and eventually iron chelation therapy to allow patients to lead a normal life. Clinically significant α-thalassemia conditions are relatively rare, but can be responsible for fatal hydrops fetalis at birth and for Hemoglobin H disease, a lifelong chronic anemia. The most frequent hemoglobin (Hb) variants in Brazil include HbS, HbC, and HbD-Los Angeles, where HbD-Los Angeles (1.6%) stands out as the third most common hemoglobin variant in the group analyzed, thus confirming the data reported by Leoneli (2001). The most frequent phenotype in the group of rare hemoglobins is Hb Hasharon in heterozygosis. This phenotype is a hemoglobin variant frequently found in populations of Mediterranean origin, thus confirming the strong European influences in the composition of the Latin American population (Huisman et al., 2001).
The primary aim was to detect the prevalence of hemoglobinopathy disorders in adult patients sent for diagnosis of anemia, which included thalassemia (HbA), (HbA2), (HbF) and sickle cell disease (HbS) in Riyadh city, Saudi Arabia. Study data were compared with other research performed in other countries. In Saudi Arabia, there are no definite data regarding the pattern of hereditary hemoglobin disorders. No screening program has been implemented in this diverse population group; therefore, this is the first study of this nature.
Methods
Sample collection
The present study was conducted in Razi Clinic Um alhammam, Riyadh Saudi Arabia. Specimens were drawn into dipotassium ethylenediaminetetraacetic acid-coated tubes (Becton Dickinson Vacutainer Systems). A total number of 329 subjects of both sexes (140 men and 189 women) were included in the study. The study was approved by the Ethical Committee Razi Clinic. Written informed consent was obtained from all the subjects included in this study.
Hemoglobin variant analysis
All specimens were analyzed in relation to the age, sex, full blood count, hemoglobin and iron profiles, and hemoglobin-electrophoresis by the Bio-Rad Variant II high-performance liquid chromatography (HPLC; Bio-Rad Laboratories) according to the manufacturer's instructions. A total of 329 subjects received from January 2006 to December 2010 for hemoglobin variant analysis were studied for various hemoglobinopathies and variants. Briefly, in this system, the samples were mixed by the Variant II sampling station, diluted with the specific hemolyzing/wash buffer, and injected into an assay-specific analytic cartridge. The Variant II dual pumps delivered a programmed buffer gradient of increasing ionic strength to the cartridge, where the hemoglobin fractions were separated based on their ionic interaction with the cartridge material. The separated hemoglobin fractions passed through a flow cell, where absorbance was measured at 415 nm; background noise was reduced with the use of a secondary wavelength at 690 nm. The raw data were integrated by the Clinical Data Management software (Bio-Rad Laboratories), and a chromatogram/sample report was generated. The integrated peaks were assigned to manufacturer-defined windows derived from the retention time, that is, the time in minutes from the sample injection to the maximum point of the elution peak, of normal hemoglobin fractions, and common variants (Fig. 1). If a peak elutes at a retention time not predefined, it is labeled as an unknown (Gupta et al., 2009).
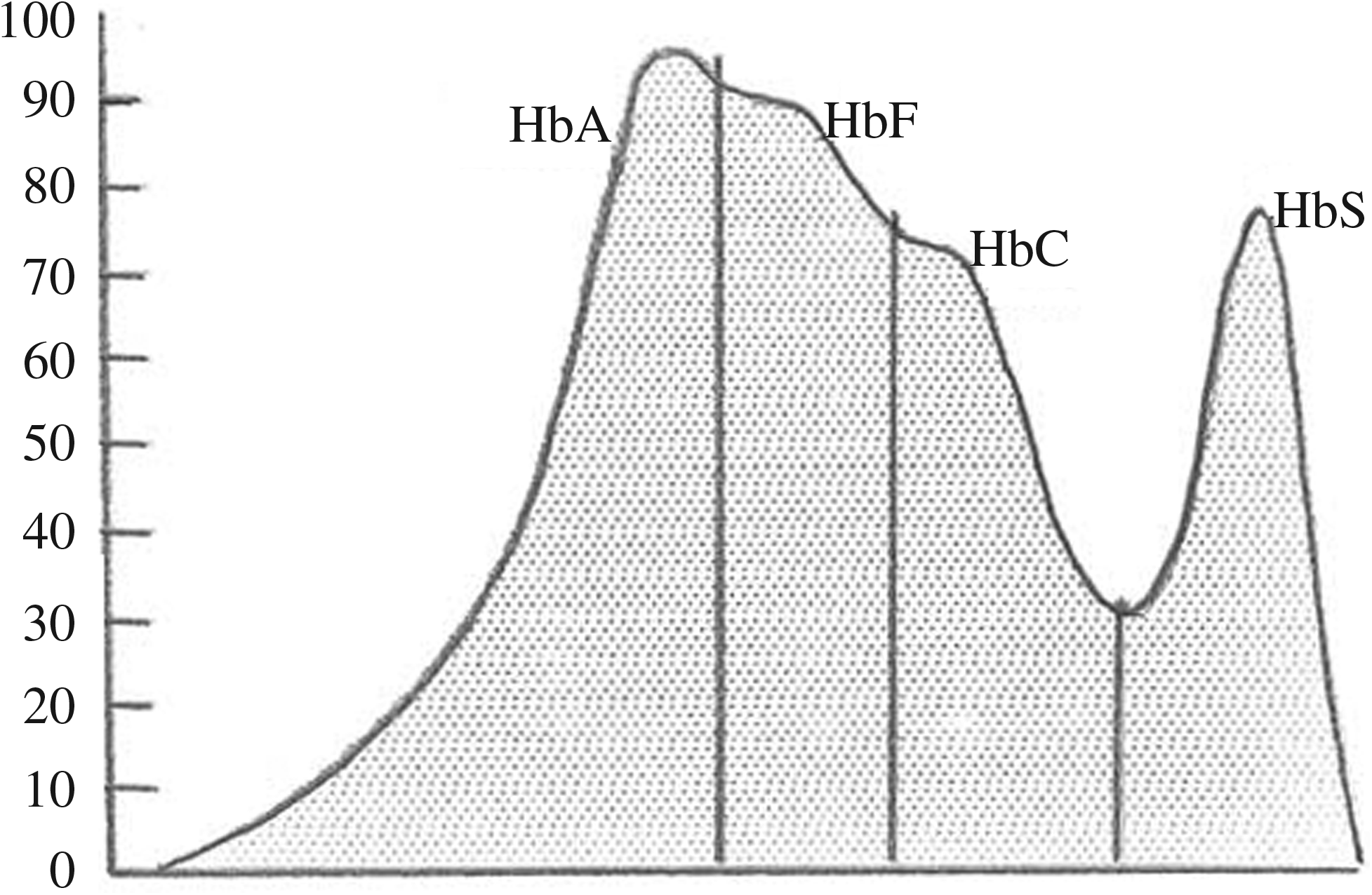
High-performance liquid chromatograms obtained on the Bio-Rad variant β-thalassemia short program for hemoglobin barts.
Statistical analysis
For the statistical analysis, SPSS v17 (PASW) was used. Paired t-test and independent sample t-tests were performed where and as required. Results were expressed as mean±SD; p-value<0.05 was considered as significant.
Results
A total of 329 adults were screened in Riyadh, Saudi Arabia, with a HPLC method. The HPLC chromatogram is diagnostic; showing a typical early elution peak representing hemoglobin Barts The chromatograms in Figure 1 show the subjects with different hemoglobin patterns. The screening showed the hemoglobin disorders such as β-thalassemic trait; increased fetal Hb (HbF), sickle cell trait, sickle cell disease, sickle cell/β-thalessamia, and normal pattern of hemoglobin-electrophereses; and iron deficiency masked thalassemia (Table 1).
Hb, hemoglobin; SD, standard deviation; TIBC, total iron-binding capacity.
Out of 329 subjects, 91 (27.7%) carried the gene for one of the conditions just described and were either affected individuals or carriers. One hundred eighteen (35.9%) subjects showed a normal pattern screen with a level of HbA (97.01%±0.4%), HbA2 (2.72%±0.55%), and an average level of HbF (0.50±0.25) as shown in Table 1. One hundred twenty (36.5% of the total) were normal with iron deficiency masked thalassemic trait on the levels of HbA2 (97.23%±0.76%) and HbA (2.67%±0.60%). Most (26.5%) were women, and 10% were men with an iron level of 3.62±2.7 μg/dL, and a total iron-binding capacity of 86.9±21.5 μg/dL. The correlation between normal electrophoresis pattern and iron deficiency masked thalassemic trait in hematological parameters is shown in Table 2, and all of the parameters show significant differences (p-value<0.001) except mean corpuscular hemoglobin concentration.
RBC, red blood cells; HCT, hematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; RDW, red blood cell distribution width.
Of the affected individuals, 33 (10.0%) were found to be affected with β-thalassemia with an elevated level of HbA2 (5.54±1.0 for men and 5.57±1.06 for women). Eleven subjects (3.3%) were affected with sickle cell and β-thalassemia disease HbSS (93.7%±1.52% for men and 64.5%±29.5% for women).
Twenty-nine patients (8.8%) were found to be sickle cell trait carriers with a level of HbS <45% (35.1%±3.59% for men and 35.26%±3.57% for women) of sickle cell disease and a slightly elevated level of HbF (0.8%±0% for men and 0.2%±0.14% for women). Thirteen patients (4.0%) had sickle cell disease with an elevated level of HbSS >45% (75.0%±11.47% for men and 68.5%±20.6% for women) and a high elevated level of HbF (20.7%±9.68% for men and 16.1%±11.5% for women). The study also illustrated 5 (1.5%) cases of men with persistence of HbF (7.12%±11.1%); fortunately, this form is less common among the population. The correlation of abnormalities of electrophoresis patterns between men and women was shown in Table 3, with no significant differences between them. Therefore, the study illustrated that it is quite a common problem and leads to a burden of health.
Discussion
Hemoglobinopathies are inherited disorders of globin, the protein component of hemoglobin. Mutations in genes coding for the globin proteins that alter protein output produce the thalassemia syndromes. Mutations in the globin genes that lead to abnormal proteins are called variant Hbs (Weatherall and Clegg, 2001) Hemoglobinopathies are the commonest genetic defect worldwide with an estimated 269 million carriers (Angastiniotis and Modell, 1998). Certain populations are particularly at risk of having a hemoglobinopathy, for example, in South East Asia, there are 90 million carriers, about 85 million in sub-Saharan Africa and 48 million in the West Pacific region (Angastiniotis and Modell, 1998).
Variant hemoglobin disorders were studied in 329 individuals, each had distinguishing characteristics with an average age of 39±6.49 years. The study illustrated that sickle cell disease and β-thalessamia are very common hemoglobinopathies found in the Saudi Arabian Population. The distribution of variant hemoglobins in these 329 subjects is shown in Table 1.
In Saudi Arabia, data regarding the hereditary hemoglobin disorder are not available so far. Therefore, the present study was designed to detect the variant hemoglobinopathy in individuals. The study revealed that normal with iron deficiency masked thalassemic trait was the most common hemoglobin found in the population, followed by β-thalassemic trait, and sickle cell disease. HbA2 and HbF levels are lower in iron deficiency than in an iron replete state. Hence, the presence of coexistent iron deficiency may mask detection of underlying β-thalassemia. If suspected, it is recommended that iron deficiency be excluded before doing quantitative HbA2 and HbF testing. Alternatively, testing should be repeated for thalassemia if the patient remains microcytic after iron stores are replaced (Zwick, 2004). Persistence of HbF, in which the patient fails to switch to HbA and continues to synthesize HbF in adult life, was found to be only 1.5%in this population. This condition has been found primarily in Greeks (Fessas and Samatoyannopoulos, 1968). Patients with persistence HbF are clinically asymptomatic. Worldwide, over 1% of couples are at risk for hemoglobin disorders (Christianson and Modell, 2004), most have at least one affected child, and most affected children die in early childhood. Although the West African death rate in children aged <5 years is 18.4%, the rate is 16.5% for children born to couples not at risk for sickle-cell disorders compared with 40% for children born to couples who are at risk (Christianson and Modell, 2004). With the high frequency of β-thalassemia, causing an increased burden on the society, it is necessary to control the incidence by effective steps. Implementation of carrier screening program offering genetic counseling and prenatal diagnosis followed by selective termination of affected cases would help in preventing the disease. Similar approaches in other countries such as Greece, Cyprus, and Sardina resulted in a marked reduction in the birth rate of affected children (Angastiniotis and Hadgiminas, 1981). Some of this population is usually asymptomatic, do not require treatment, and lead a reasonably good quality of life but they are dangerous because of the possibility of homozygous or double heterozygous inheritance through marriage of unaware couples or silent spread as a trait. This is a serious health threat to our nation, if it is allowed to continue without taking measures for prevention.
Conclusion
In the present study, we have at least reached a concrete conclusion that hereditary hemoglobin disorders are very common in Saudi Arabia, on which the health authorities should focus. Awareness has to be created at the national level to reduce the incidence of hereditary hemoglobin disorders in the community. It is mandatory to detect the trait in the general population, and large-scale and proper genetic counseling should be ensured. It is time to think about establishing strategies for molecular and prenatal diagnosis to prevent the further spread of the disease.
Footnotes
Acknowledgments
The author convey their sincere thanks to the patients who participated in this study. The author thanks the following staff from Alrazi Clinic: Dr. Sami Mustafa, Medical Director, Dr. Badawi Radwn, and Dr. Adell Awad for their assistance and support with this study; they extend their gratitude to Prof. Talal Bakery and Dr. Mohammed A. Alsaeed for proof reading and also for his valuable comments in improving this article; they are grateful to the technicians Safeyah Sulog and Shareen Soltan.
Disclosure Statement
No competing financial interests exist.