
Abstract
Congenital heart disease (CHD) is one of most common birth defects threatening newborns' health. Over the past few decades, a variety of CHD-causing gene mutations have been identified, but the pathogenic mechanism of congenital heart disease is yet not very clear. The aim of this study was to identify potential pathologic mutations in the NODAL gene and to gain insight into the etiology of CHD. By using amplification with polymerase chain reaction and sequence analysis of NODAL in 800 patients with nonsyndromic CHD and 250 healthy controls, we identified 3 nonsynonymous variants. One of them was first identified in the present study. These variants were not observed in 250 controls. To our knowledge, this is the first study to suggest that NODAL may be involved in the etiology of nonsyndromic CHD in a Chinese population.
Introduction
C
Related genetic studies revealed that most of these genes encode transcription factors that regulate specific events in heart development, such as ventricular septation or outflow tract morphogenesis (Bruneau, 2008). The first identified single-gene mutation was in the T-box transcription factor gene TBX5, which predominantly affects atrial septal defects (ASDs), ventricular septal defects (VSDs) and conduction-system defects (Basson et al., 1997).
Located in 10q22.1 and containing 3 exons, NODAL is a member of the transforming growth factor (TGF)-β gene family and is expressed during mouse gastrulation; it is also an important morphogen and regulator of cell fate in both embryologic and adult systems (Zhou et al., 1993; Schier, 2003). TGF-β/activin/NODAL signaling has been demonstrated to play a role in maintaining pluripotency in human embryonic stem cells because disruption of this signaling pathway results in their differentiation. NODAL also controls the direction of cardiac looping and the development of left-to-right asymmetry in chickens and mice (Olson and Srivastava, 1996). NODAL has been proposed to contribute to cardiac development and may therefore lead to CHD.
We hypothesize that NODAL may contribute to the development of CHD. Our study aimed to identify potential pathogenic mutations for NODAL in 800 Chinese children with CHD and to provide insights into the etiology of CHD.
Materials and Methods
Study population
A total of 800 patients with nonsyndromic CHD and 250 controls with no reported cardiac phenotype were recruited for this study from Lanzhou University. The phenotype of CHD included VSD, pulmonary atresia or stenosis, tetralogy of Fallot, double-outlet right ventricle, ASD, patent ductus arteriosus, aortic coarctation, pulmonary hypertension, and other complex cardiac malformations (Table 1). Informed consent was obtained from patients' parents or guardians. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Ethics Committee of the National Research Institute for Family Planning.
Clinical assessment of the patients included anthropometric measurement, physical examination for dysmorphism and malformation, and radiologic evaluation. The patients also underwent chest radiography, electrocardiography, and ultrasonic echocardiography.
DNA analysis
Genomic DNA was extracted from peripheral blood leukocytes by using standard methods. The human NODAL gene is located on 10q22.1 and is encoded by 3 exons. Three exons and nearby introns of NODAL were amplified by polymerase chain reaction (PCR) using 3 pairs of NODAL gene-specific primers (Table 2). PCR products were sequenced using the appropriate PCR primers and the BigDye Terminator Cycle Sequencing kit (Applied Biosystems, Foster City, CA) and run on an automated sequencer, ABI 3730XL (Applied Biosystems) to perform mutational analysis.
Results
Sequence analysis of NODAL in 800 patients with nonsyndromic CHD identified 3 nonsynonymous variants, 1 common CHD-causing mutation, and a missense variation rs10999334 (Table 3); 1 was first identified by the present study. One of the novel nonsynonymous variants (c. T182A p. Leu61Asn;) was located at the TGFb_prope domain. The other novel mutation, p. Glu203Lys, was located between the TGFb_prope and the TGFb domain.
ASD, atrial septal defect; PDA, patent ductus arteriosus; PH, pulmonary hypertension; VSD, ventricular septal defect.
The variant c. T182A and G607A resulted in substitutions of leucine for asparagine at position 61 and glutamic acid for lysine at position 203, respectively. Leu61 and Glu203 were highly conserved among many species (human, mouse and rat, shown in Fig. 1).

Conservation and location of the variants found in NODAL. Three nonsynonymous variants (L61N, H165R, E203K) are indicated by black arrows. The conservative test was performed between human NODAL, mouse NODAL, and rat NODAL. The transforming growth factor-β propeptide domain is highlighted by black lines.
We also identified some nonpathogenic variants: rs2279253 and rs7909303. An association analysis compared these single-nucleotide polymorphisms (SNPs) between patients with CHD and 250 healthy controls but did not show statistically significant differences.
Discussion
During early development of vertebrate embryos, specification of mesoderm and endoderm germ layers requires the Nodal/Activin signaling pathway. NODAL signaling activates a canonical TGF-β pathway involving activin receptors, Smad2 transcription factors, and FoxH1 coactivators. In addition, NODAL signaling depends on co-receptors of the EGF-CFC family and antagonized by the Lefty and Cerberus families of secreted factors (Schier and Shen, 2000). EGF-CFC genes encode a group of structurally related proteins that play key roles in intercellular signaling pathways during vertebrate embryogenesis. This multigene family includes Xenopus FRL-1, zebrafish one-eyed-pinhead (oep), mouse cripto (Cr-1) and cryptic, and human cripto (CR-1) and criptin. The NODAL gene is expressed in the left lateral plate mesoderm and activates the Nodal pathway within the left side of the heart (Long et al., 2003). NODAL hypomorphic mutant mice form abnormal hearts; studies in zebra fish embryos deficient for zygotic NODAL co-receptor have also been shown to develop abnormal hearts (Gritsman et al., 1999).
The number of Han Chinese samples in this study is consistent with numbers in the HapMap project (www.hapmap.org; approximately 82 individuals), and this study empirically has more depth. In a previous study, missense mutations of NODAL were discovered in patients with classic heterotaxy and looping cardiovascular malformations (Mohapatra et al., 2009). In this study of Chinese children with CHD, 1 novel nonsynonymous variant within the coding region of NODAL was discovered.
This mutation L61N, located in the preprotein structural region, functions through the disulfide bond of its dipolymer to connect the TGF-β-conjugated protein. In the SNP found in this study, a nonpolar amino acid leucine was replaced by 1 polar amino acid asparagine. The change may in turn result in a change of protein structure. On the other hand, TGF-β dipolymer may affect its connection with TGF-β-conjugated protein. PolyPhen (http://genetics.bwh.harvard.edu/pph/), a tool that predicts the possible effect of an amino acid substitution on the structure and function of a human protein, predicted that the change of this amino acid would probably damage the protein structure; in addition, the site was conserved among species. The ExPASY ProtScale (www.expasy.org/cgi-bin/protscale.pl), which is defined by a numeric value assigned to each type of amino acid to predict the hydrophobicity or hydrophilicity scales and the secondary structure conformational parameter scales (and many other scales based on different chemical and physical properties of the amino acids), indicated that the hydropathicity of the L61N mutant protein was very different from that of the wild type (Fig. 2). Functional studies should be performed to explore the biological mechanism and pathogenicity of the L61N alternative protein.

The difference between the hydrophilicity of the wild-type protein
In addition, the missense variation E203K was located between the domain of precursor and mature proteins and led to a change from a polarity-negative glutamic acid into a nonpolar, neutral lysine. As predicted by the ExPASY ProtScale, the hydropathicity of the E203K variation protein was little different from that of the wild type (Fig. 3). This alteration occurs in only 0.017 of the Chinese population, according to the HapMap project. In our study, the prevalence was 1/800 (0.00125).
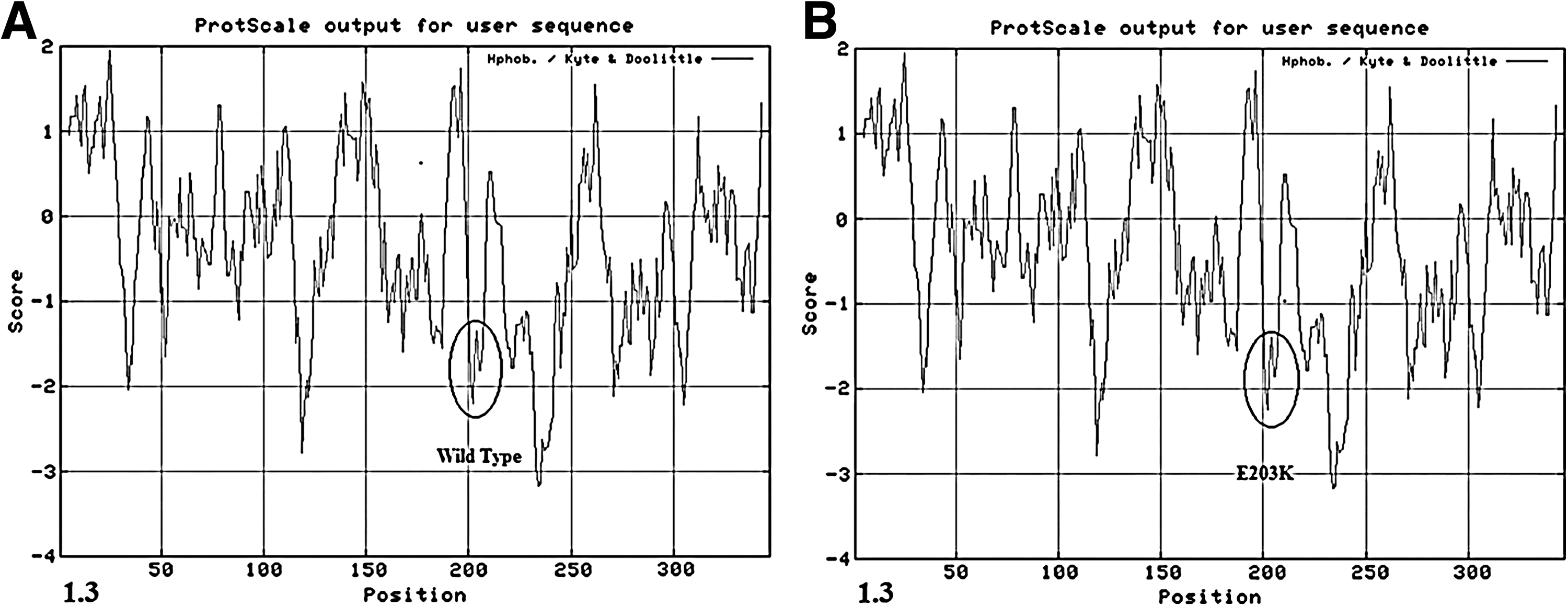
The difference between the hydrophilicity of the wild-type protein
In a previous study (Roessler et al., 2009), 16 CHD-causing mutations were found in a cohort of 375 unrelated individuals prospectively ascertained at several urban centers. Those participants had a wide spectrum of defects, including transposition of the great arteries, tetralogy of Fallot, double-outlet right ventricle, ASD, common atrioventricular canal, and interrupted aortic arch but did not contained VSD. Compared with Roessler and colleagues' study, the present study identified 3 pathogenic nonsynonymous variants of the NODAL coding region among 800 Chinese patients with CHD. The mutation rate of the NODAL gene was lower, and only 1 of those 3 mutations (H165R) overlapped with the earlier study. We found only 1 H165R mutation in 1 of the 253 patients with ASD; the estimate of the incidence of genetic variation is probably underestimated because of the case distribution. The data possibly reflected the obvious ethnic differences between the Caucasus and the Chinese population. Meanwhile, 9 mutations found in Roessler and colleagues' study were in patients with tetralogy of Fallot, and the patient ratio of this subtype was 6.5% in the present study. This finding could suggest that mutations in the NODAL gene were more likely to cause tetralogy of Fallot. The CHD subtype structures of Roessler and colleagues' study lacked the VSD subtype, which was the most frequent in the present study. Furthermore, VSD is the most common form of CHD in both children and adults, occurring in 50% of all children with congenital heart defect and in 20% as an isolated lesion (Moller et al., 1995). In the present study we only found 1 mutation in VSD patients, who made up 55% of the patient cohort. We may be the first to have identified a CHD-causing mutation in a VSD patient; the result could extend the study of the NODAL gene and the etiology of nonsyndromic CHD, especially in VSD.
In conclusion, the mutational analysis of NODAL in 800 Chinese patients with CHD revealed 2 novel nonsynonymous variants. To our knowledge, our study suggests for the first time that NODAL may be involved in the etiology of nonsyndromic CHD in the Chinese population and is the first to identify a potential pathogenic mutation in the NODAL gene in patients with VSD.
Footnotes
Acknowledgments
Lei Sun and Longfei Cheng drafted the manuscript and carried out the molecular genetic studies. Hanquan Dong, Jing Wang, Yuan Mu, Adong Shen, and Xiaotian Li collected all samples and participated in the design of the study. Binbin Wang, Guoying Huang, Zhongzhi Li, and Xiaodong Xie participated in manuscript revision and samples collection. Hui Li and Xu Ma conceived the study, participated in its design and coordination, and helped to draft the manuscript.
This work was supported by the National Basic Research Program of China (2010CB529504) and the National Science & Technology Pillar Program of China (No.2008BAH24B05)
Disclosure Statement
The authors declare that no competing financial interests exist.