
Abstract
Aim: Spinal muscular atrophy (SMA) is a common autosomal recessive neuromuscular disorder. It is caused by mutations in the SMN1, and its clinical severity is modified by copy number variations of the SMN2. According to previous studies, deletion of SMN1 exon 7 is the most frequently observed in patients with SMA. Therefore, molecular analyses exploiting this genetic lesion could be beneficial in the diagnosis of SMA. Unfortunately, in many geographical regions, physicians do not have the latest molecular screening technologies at their immediate disposal. Thus, to overcome this issue, we developed an SMA-diagnosing system using dried blood spots (DBS) placed on filter paper to facilitate remote diagnosis. Methods: In this study, we validate the applicability of DBS on Flinders Technology Associates (FTA) filter paper for detecting SMN1 exon 7 deletions and copy number variations of SMN1 and SMN2. To detect exon 7 deletions in SMN1, polymerase chain reaction (PCR)-restriction fragment length polymorphism analysis was conducted by using DNA extracted from the DBS on FTA filter paper that had been stored at room temperature for a period of up to 4 years. To determine the copy numbers of SMN1 and SMN2, we carried out SYBR green-based real-time PCR by using the same blood specimens. Results: The results obtained from the DBS on FTA filter paper were in complete concordance with those analyses using fresh blood specimens. This indicates that DBS on filter papers is a reliable method for SMA patient detection and carrier screenings. Conclusion: The SMA-diagnosing system, combined with the mailing of DBS on filter paper, will be beneficial for patients suffering from neuromuscular disorders in areas with limited or no access to diagnostic facilities with molecular capabilities.
Introduction
S
The gene responsible for SMA is the survival motor neuron (SMN) gene. This gene has two nearly identical copies, SMN1 and SMN2 (Lefebvre et al., 1995). It has been shown that SMA is caused by a homozygous disruption of SMN1, and further, the clinical severity of the disease is modified by copy number variations of SMN2 (Feldkötter et al., 2002). To a limited degree, the presence of SMN2 copies can compensate for the deletion of SMN1, and it has been shown that in the absence of SMN1, increased copy numbers of SMN2 can improve the clinical severity of the disease (Harada et al., 2002). Additionally, the presence of the neuronal apoptosis inhibitory protein (NAIP) gene is also associated with the clinical severity (Glotov et al., 2001).
Most cases of SMA present with the complete absence of SMN1 (Parsons et al., 1998). Patients with an SMA-like presentation should be tested for the presence of a homozygous deletion of SMN1 by using methods that are 98.4%-99% sensitive and 98.3%-100% specific (Beck et al., 2001; Mailman et al., 2002). Therefore, the detection of SMN1 deletions is highly beneficial in diagnosing SMA. Additionally, copy number analysis of SMN1 may also be valuable for carrier testing, because all carriers have only 1 copy of SMN1.
When discussing the topic of molecular testing, it is essential to note that the means to conduct molecular analysis is not universally available in all geographical regions of the world. With this in mind, if a simple method of collection, storage, and shipment of samples could be established, then all patients can gain access to the benefits of molecular analysis regardless of geographical or industrial limitations. This is true not only for SMA but also for other inherited disorders as well (Aggarwal et al., 1992). In this study, we proposed a novel SMA-diagnosing system using dried blood spot (DBS) on filter paper, which effectively facilitates the simplified collection, storage, and shipment of patient samples to centralized testing facilities.
Patients and Methods
Patients
ID-1 (woman, SMA type 1). She was referred to Kobe University Hospital for the evaluation of muscle weakness at 1 month old. She did not obtain head control or sit by 3 years.
ID-2 (woman, SMA type II). She was referred to Kobe University Hospital for the evaluation of muscle weakness at 6 months old. She obtained head control at 4 months old, but she could not sit without aid. She underwent tracheostomy at 2 years of age.
ID-3 (woman, SMA type III). She was referred to Kobe University Hospital for the evaluation of muscle weakness affecting dominantly lower limbs at 12 years old. Her gait disturbance had been noticed since she was 3 years old.
ID-4 (woman, SMA type I). She was referred to Kobe University Hospital for the evaluation of muscle weakness at 1 year 5 months old. She did not obtain head control or sit without aid.
ID-5 (man, SMA type III). He was referred to Kobe University Hospital for the evaluation of muscle weakness affecting dominantly lower limbs at 5 years of age.
Before the molecular analysis, informed consent was obtained from the parents of the patients. This study was approved by the Ethical Committee in Kobe University.
Extraction of DNA from Flinders Technology Associate elute microcard™
For the collection of blood samples from SMA suspicious patients, we adopted the use of FTA® Elute Cards (Whatman® Inc, Schleicher & Schuell, Clifton, NJ, art no. WB 120410). Storage periods of the Flinders Technology Associate (FTA) Elute Cards varied from 1 to 4 years. All FTA Elute Cards were stored at room temperature. To extract DNA from the DBS, 5 circles were punched out from the center of a blood spot by using a Φ3 mm hole puncher, as shown in Figure 1. The punched circles were put into a sterile 1.5 mL microfuge tube and washed by adding 500 μL of sterile water and 3 cycles of pulse vortexing. Excess water used for washing was removed from the tube by using a sterile pipette. After this step, the circles obtained were further centrifuged for 5 s, and excess water was removed again by pipetting. After washing, these punched circles were completely immersed in 100 μL of TE buffer, used as an eluant followed by heating at 95°C for 30 min. At the end of the incubation period, the tubes were pulse vortexed ∼60 times. The eluant was stored at −20°C until being used as a DNA source for further examination (Whatman, Inc., Clifton, NJ).

Image of FTA Elute microcard™ after being punched. On filter paper, five holes in the center of one well can be observed. To avoid cross contamination between samples, a micropunch was carried out thrice on a clean piece of filter paper followed by wiping of the holepuncher with 95% ethanol. This was done between all FTA samples. FTA, Flinders Technology Associates.
Deletion test
Polymerase chain reaction (PCR) was performed according to the methods described by van der Steege et al. (1995). For each reaction, 300-500 ng of DNA was used. The oligonucleotide primers for exon 7 in the SMN1 and SMN2 were R111 (Lefebvre et al., 1995) and×7-Dra (van der Steege et al., 1995), and those applied for use on exon 8 of both genes were 541C950 (Lefebvre et al., 1995) and 541C1120 (Lefebvre et al., 1995). To discriminate between SMN1 and SMN2 products, PCR amplicons were completely digested with DraI (Takara Biomedical, Tokyo, Japan) for exon 7 and DdeI (Takara Biomedicals) for exon 8. The restriction enzymes DraI and DdeI only cleave the amplified fragments from SMN2 exon 7 and 8, respectively. PCR amplification of the NAIP-specific sequence, exon 5, was performed according to the method reported by Roy et al. (1995).
Sequencing analysis
To identify and confirm the presence of hybrid genes, direct sequencing analysis was performed by using a BigDye Terminator V3.0 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and a genetic analyzer (ABI Prism 310; Applied Biosystems), with DNA Sequencing Analysis Software (Applied Biosystems). PCR amplicons were used as templates along with the forward primer of intron 6 and reverse primer of exon 8 for sequencing reactions.
Copy number analysis
For determination of SMN1 and SMN2 copy numbers, we used a quantification method utilizing real-time PCR and a LightCycler instrument (Roche Diagnostics, Mannheim, Germany). SMN1 exon 7 was amplified with a primer set of telSMNex7forw (5′-TTT ATT TTC CTT ACA GGG TTT C-3′) (Feldkötter et al., 2002) and telSMNint7rev (5′-GTG AAA GTA TGT TTC TTC CAC GTA-3′) (Feldkötter et al., 2002). SMN2 exon 7 was amplified with a primer set of cenSMNex7forw (5′-TTT ATT TTC CTT ACA GGG TTT TA-3′) (Feldkötter et al., 2002) and cenSMNint7rev (5′-GTG AAA GTA TGT TTC TTC CAC GCA-3′) (Feldkötter et al., 2002). The CFTR was used as a reference gene for the relative quantification of SMN1 and SMN2. CFTR was amplified with a primer set of CF621F (5′-AGT CAC CAA AGC AGT ACA GC-3′) (McAndrew et al., 1997) and CF621R (5′-GGG CCT GTG CAA GGA ATG TTA-3′) (McAndrew et al., 1997). The details of the experimental procedure we applied have been described elsewhere (Tran et al., 2008).
Statistical analysis
To evaluate the performance of the FTA Elute Microcard method described in this study, sensitivity and specificity were calculated by using standard statistical analyses.
Results
DNA concentration or recovery of DNA from the DBS on FTA elute microcard
To assess the performance of FTA Elute microcards (Whatman Inc, Schleicher & Schuell, art no. WB 120410), we selected cards based on their storage time (Table 1). The storage time of the individual cards varied from 1 to 4 years. Concentration of DNA extracted from the cards ranged between 66.5 and 321.3 mg/L (mean 204.4 mg/L). DNA samples had OD 260: OD 280 values of 1.56 and 1.86, respectively. The concentration and purity of the isolated DNA was sufficient for subsequent PCR analysis.
Sample was prepared from five punched circles that had been (Φ3 mm) incubated in 100 μL of TE buffer.
Deletion test
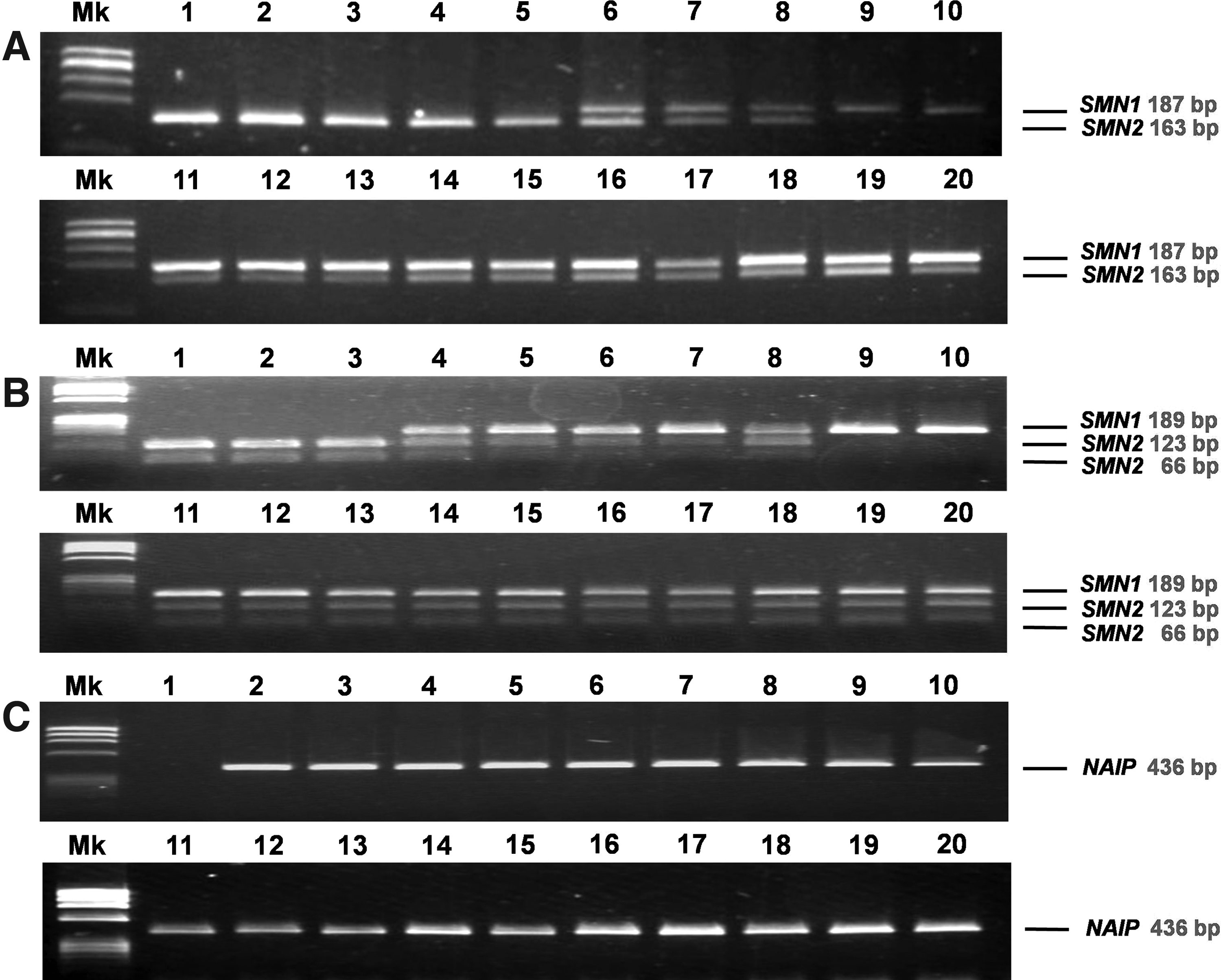
To determine the presence and/or absence of SMN exon 7, we performed a PCR-enzyme digestion analysis of these exons by using DNA samples extracted from the FTA Elute microcards. After complete digestion by DraI, nondigested PCR products from SMN1 exon 7 (187 bp) could not be observed in five samples (lanes 1-5), although these samples clearly show the presence of digested PCR products from SMN2 exon 7 (163 bp) (Fig. 2). In 13 samples (lanes 6-8, 11-20), nondigested PCR products from SMN1 exon 7 were present along with the digested PCR products from SMN2 exon 7. In two samples (lanes 9 and 10), the presence of nondigested PCR products from SMN1 exon 7 SMN1 were confirmed, but digested PCR products from SMN2 exon 7 could not be observed. These data obtained from analyses using DNA extracted from DBS on the FTA Elute microcards were compatible with those obtained from analyses using DNA prepared from EDTA-blood (Table 2). In the current study, we compared and evaluated the results of the SMN1 exon 7 deletion test by using DNA extracted from DBS on the FTA Elute Microcard with test results obtained from DNA prepared from EDTA-blood. Both sensitivity and specificity displayed outstanding results at 100% (Table 3). Data obtained from analyses using DNA prepared from EDTA-blood are not shown in this article.
These data obtained from analyses using DNA extracted from DBS on the FTA Elute microcards were compatible with those obtained from analyses using DNA prepared from EDTA-blood. (The data of SMN-deletion test using DNA prepared from EDTA-blood not shown).
The deletion test using card blood was completely compatible with the freshly prepared sample.
DBS, dried blood spot; FTA, Flinders Technology Associates; CNR, calibrator-normalized ratio; del, deletion; nondel, nondeletion.
Sensitivity test=5/(5+0)×100%=100%.
Specificity test=15/(15+0)×100%=100%.
From Fisher analysis, exact Fisher p was 0.72. It indicated a nonsignificant difference between the result from DBS on filter paper and EDTA-blood.
Similar—but not identical—results were obtained from the analysis conducted on SMN exon 8. After complete digestion by DdeI, three samples (lanes 1-3) did not have nondigested PCR products from SMN1 exon 8 (189 bp), although the presence of two digested PCR products from SMN2 exon 8 (123 bp, 66 bp) could be detected (Fig. 2). Fifteen samples (lanes 4-8, 11-20) showed the presence of nondigested PCR products from SMN1 exon 8 and two digested PCR products from SMN2 exon 8. Two samples (lanes 9 and 10) showed the presence of a nondigested PCR product from SMN1 exon 8 but did not show digested PCR products from SMN2 exon 8. These two samples were taken from healthy controls. The results obtained using DNA extracted from DBS on the FTA Elute microcards were compatible with those obtained from DNA prepared from EDTA-blood.
Interestingly, two samples (lanes 4 and 5) were absent for SMN1 exon 7, whereas the presence of SMN1 exon 8 could be readily confirmed. These samples were taken from patients with the so-called hybrid SMN genes (Figs. 2 and 3B). To confirm the initial results suggesting the presence of a hybrid SMN gene in DNA samples extracted from DBS on the FTA Elute microcard, we performed a sequence analysis of the PCR product that spanned intron 6, exon 7, intron 7, and exon 8. In the sequence between intron 6 and intron 7, three SMN2 specific nucleotides were present. In stark contrast, in exon 8, both SMN1 and SMN2 specific nucleotides were present in the sequence (Fig. 3A).

In addition, we performed PCR analysis on NAIP exon 5. Only 1 out of 20 samples showed the absence of NAIP exon 5 (lane 1) (Fig. 2). The sample was obtained from a patient with type I SMA. This result was compatible with a previous result obtained using DNA prepared from EDTA-blood (data not shown).
Copy number analysis
Copy number variations in SMN1 and SMN2 were determined by a relative quantification method based on the calibrator-normalized ratios (CNR) (Tran et al., 2008). When DNA samples showed CNR values of 0.6-1.4, 1.5-2.4, 2.5-3.4, and 3.5-4.4, it was considered that they carried 1, 2, 3, and 4 copies of the gene, respectively. Copy numbers determined from CNR values obtained for the DBS samples were consistent with those determined for EDTA-blood samples.
In this study, SMN1 copy number variations in patients with SMA (ID 1-5) was not a major issue, because they had been determined through previous deletion studies that there was a deletion of SMN1. For these samples, we only conducted copy number analyses for SMN2. In these patients, SMN2 copy number varied from two to four copies; two copies in Type I, three copies in Type I, II, and III, and four copies in Type III. The results displayed a tendency that SMN2 copies had a more profound effect on the phenotype (Table 2).
In the present study, 3 out of 15 non-SMA samples (ID 6-8) carried a single copy of SMN1. These samples belong to the parents of patients with SMA. Consequently, the parents were also diagnosed as with carrier status of SMA at the molecular genetic level, as well as at the clinical level.
We also examined SMN1 and SMN2 copy number variations in 12 controls (ID 9-20). The samples were obtained from healthy persons. With the exception of one sample (ID 20), all control samples had two copies of SMN1 (Table 2). Sample ID 20, on the other hand, had three copies of SMN1. The presence of SMN2 in control samples varied from 0 to 3 copies.
Discussion
We demonstrated the applicability of DBS on filter paper for molecular diagnostic analysis of SMA. In this study, a simple boiling method was applied for DNA extraction from DBS affixed on five Φ3 mm-circles of filter paper. This amount corresponds to ∼50 μL of whole blood, and the method has been adapted from a method previously described (Kogan et al., 1987; Jinks et al., 1989). The total amount of DNA extracted from five Φ3 mm-circles was between 6.65 and 32.13 μg. The OD260/OD280 ratio of the DNA extracted from DBS was 1.56-1.86 (Table 1), which retained enough purity for subsequent PCR analysis. Without exception, in all samples, we successfully amplified the target genes: SMN1, SMN2, and NAIP. To determine the hybrid SMN gene, we amplified a 1011 bp-fragment encompassing intron 6 to exon 8 in the SMN1 or SMN2 genes from DBS. Similarly, Chaisomchit et al. (2005) amplified a 1039 bp-fragment of the β-globin gene from DBS on filter paper stored for 11 years, thus demonstrating the remarkable stability of DNA in DBS affixed on filter paper.
By conducting PCR using DNA derived from DBS on filter paper, we were successful in demonstrating the existence or absence of SMN1, SMN2, and NAIP. PCR-enzyme digestion experiments in this study were performed according to the method described by van der Steege et al. (1995). To determine the copy numbers of the SMN genes, we conducted a set of quantitative real-time PCR studies according to previously described methods (Tran et al., 2008). All data obtained from DBS on filter paper experiments were compatible with those obtained using EDTA-blood samples.
Majumdar et al. (2005) used multiplex-PCR to detect zero copies of SMN1 in patients with SMA and a single copy of SMN1 in carriers. Their results, combined with the results reported in this study, collectively indicate that the molecular diagnosis of SMA using EDTA-blood samples can be replaced by DBS on filter paper. Wijnen et al. (2008) sums up the potential advantages of genotyping with DBS as follows: (1) no phlebotomist is necessary, (2) genotyping results are known when the patient visits the clinician, and the clinician can take these results into account when he/she prescribes (other) drugs, (3) transport is easy, and there is a reduction in transport costs, because only the cost of an envelope and filter paper are incurred, and (4) DNA extraction from DBS is rapid, simple, and expensive DNA isolation kits are unnecessary. Accurate diagnosis based on molecular analysis can be simply obtained by mailing the filter paper with a DBS affixed to a central diagnostic center.
In conclusion, the evolution of molecular diagnostic studies have been ground breaking rather than incremental; however, the consideration of how these novel techniques can be utilized to exploit their full range of effectiveness has not been granted ample attention. In this study, we showed that DNA samples isolated from DBS serve as a satisfactory source for subsequent SMA molecular diagnosis even after 4 years of storage at room temperature. An SMA-diagnosing system, combined with the mailing of DBS on filter paper, will be beneficial for patients suffering from neuromuscular disorders in areas with limited or no access to diagnostic facilities with molecular capabilities.
Footnotes
Acknowledgments
This work was supported by a Grant-in-Aid from the Ministry of Education, Science, Sports, and Culture of Japan and a Grant-in-Aid from the Research Committee of SMA, the Ministry of Health, Labour, and Welfare of Japan.
Disclosure Statement
No competing financial interests exist.