
Abstract
Aims: Most individuals with intellectual disability and/or autism are tested for Fragile X syndrome at some point in their lifetime. Greater than 99% of individuals with Fragile X have an expanded CGG trinucleotide repeat motif in the promoter region of the FMR1 gene, and diagnostic testing involves determining the size of the CGG repeat as well as methylation status when an expansion is present. Results: Using a previously described triplet repeat-primed polymerase chain reaction, we have performed additional validation studies using two cohorts with previous diagnostic testing results available for comparison purposes. The first cohort (n=88) consisted of both males and females and had a high percentage of abnormal samples, while the second cohort (n=624) consisted of only females and was not enriched for expansion mutations. Data from each cohort were completely concordant with the results previously obtained during the course of diagnostic testing. Conclusions
Introduction
F
Historically, the detection of FMR1 trinucleotide expansions has utilized a combination of polymerase chain reaction (PCR) and Southern blotting. These two methods are routinely used in laboratories throughout the world and provide complementary information needed to make an accurate diagnosis. One of the biggest pitfalls related to testing with these standard molecular methods is the labor-intensive nature of the Southern blot analysis. For many diagnostic laboratories, it often takes a week or longer for Southern blots to be performed and interpreted. Additionally, most still use radioisotope-labeled probes given their proven history of performance and reliability. Since the inception of diagnostic testing for Fragile X, different testing strategies to facilitate the move away from Southern blots have been met with limited success. The ability to rapidly identify trinucleotide expansions in an efficient and cost-effective manner has been revisited in recent years and it now appears that several very similar protocols to address this challenge have been developed (Tassone et al., 2008; Chen et al., 2010; Filipovic-Sadic et al., 2010; Hantash et al., 2010; Lyon et al., 2010). Each of these new methods relies on a triplet repeat-primed PCR to separate those individuals with apparent CGG expansions from those without expansions. Typically, this is based on a threshold of 55 trinucleotide repeats; however, the threshold can be adjusted according to the needs of the user. This considerable advancement in testing allows the opportunity to significantly reduce the number of Southern blots needed in most diagnostic settings.
In this report, we describe the clinical validation of the laboratory-developed FMR1 PCR assay, a previously reported triplet repeat-primed PCR method (Lyon et al., 2010), in a cohort highly enriched for expansions of various sizes in both male and female samples. Given the greater challenge of identifying trinucleotide expansions in females, we subsequently tested the success of this rapid PCR assay in a substantially larger female-only cohort. Using samples that were previously tested in a diagnostic setting by both Southern analysis and standard fluorescent PCR, we have demonstrated complete concordance in both validation cohorts. To further investigate the sensitivity of the assay, we generated synthetic female samples by mixing DNA specimens from males with differing allele sizes. Here again, the assay was able to identify samples with expansions>55 repeats, even in the presence of high normal alleles that are considered to have the potential to mask the expansion. Given the successful validation of the laboratory-developed triplet repeat-primed FMR1 PCR assay by our laboratory, we have now integrated the rapid PCR-based assay into our routine workflow for all diagnostic samples referred for Fragile X analysis.
Materials and Methods
Validation samples
Samples from patients previously tested for Fragile X were used in the validation of the laboratory-developed triplet repeat-primed FMR1 PCR assay. The first cohort consisted of 88 samples (a mixture of male and female samples) and had a high percentage of samples with expanded alleles. The second female-only cohort consisted of 624 samples. Molecular testing results for all 712 samples were previously determined during routine diagnostic testing by the Molecular Diagnostic Laboratory of the Greenwood Genetic Center using concurrent Southern and fluorescent PCR analyses. All DNA samples were diluted to ∼10 ng/μL before PCR amplification using the laboratory-developed FMR1 PCR assay.
Molecular analysis used for the validation of the triplet repeat-primed FMR1 PCR assay
Validation samples were analyzed using a validated, lab-developed FMR1 PCR assay utilizing analyte-specific reagents (ASR) and general purpose reagents (GPR) obtained from Celera Corporation (Alameda, CA). These reagents are commercially available (distributed by Abbott Molecular, Des Plaines, IL) and the validation assays were performed as described in detail below.
Triplet repeat-primed FMR1 PCR assay
A triplet repeat-primed PCR-based assay that amplifies CGG repeats in the FMR1 gene was used as previously described (Lyon et al., 2010). Briefly, a pair of PCR primers generates different sized amplicons depending on the size of the CGG repeat region. The forward PCR primer is located upstream of the FMR1 CGG region. The fluorescently labeled reverse primer binds inside the CGG region and contains an eight-nucleotide-long tail. PCRs (20 μL) were set up as follows: 13 μL High GC PCR Buffer, 1.2 μL TR PCR Enzyme Mix, 0.8 μL FMR1 Primers-2, 2 μL water, and 3 μL of DNA template (10 ng/μL). PCRs were prepared on ice or on cold blocks. PCR was performed on an ABI GeneAmp® PCR System 9700 thermal cycler (Applied Biosystems, Foster City, CA) with cycling conditions of 98.5°C for 30 s, 53°C for 30 s, and 75°C for 60 s for 50 cycles followed by a hold at 4°C. Fifty cycles of PCR allow a sufficient amount of PCR product for high fluorescent signals, thus increasing robustness for clinical testing from whole blood and dried blood spots.
Capillary electrophoresis analysis
Twenty microliters of Hi-Di™ Formamide (Applied Biosystems) and 2 μL of ROX 1000 Size Standard (Celera Corporation) were combined with 2 μL PCR product. Samples were denatured at 95°C for 2 min before loading onto an Applied Biosystems 3730xl DNA analyzer with POP-7™ polymer. Data from the run were visualized using GeneMapper v.4.0 (Applied Biosystems) software. A threshold of 55 CGG repeats was set to distinguish between normal and expanded FMR1 alleles. The size (in bp) of the threshold was calculated using the following formula: #CGG repeats=(peak size in base pairs−134)/3, which is based on the primer binding sites and the number of base pairs in the amplicon excluding CGG repeats. The sample was considered normal if the CGG repeat ladder in the electropherogram did not pass the threshold (<299 base pairs). The sample was identified as having an expanded allele if the CGG repeat ladder crossed the threshold (≥299 base pairs; Fig. 1).
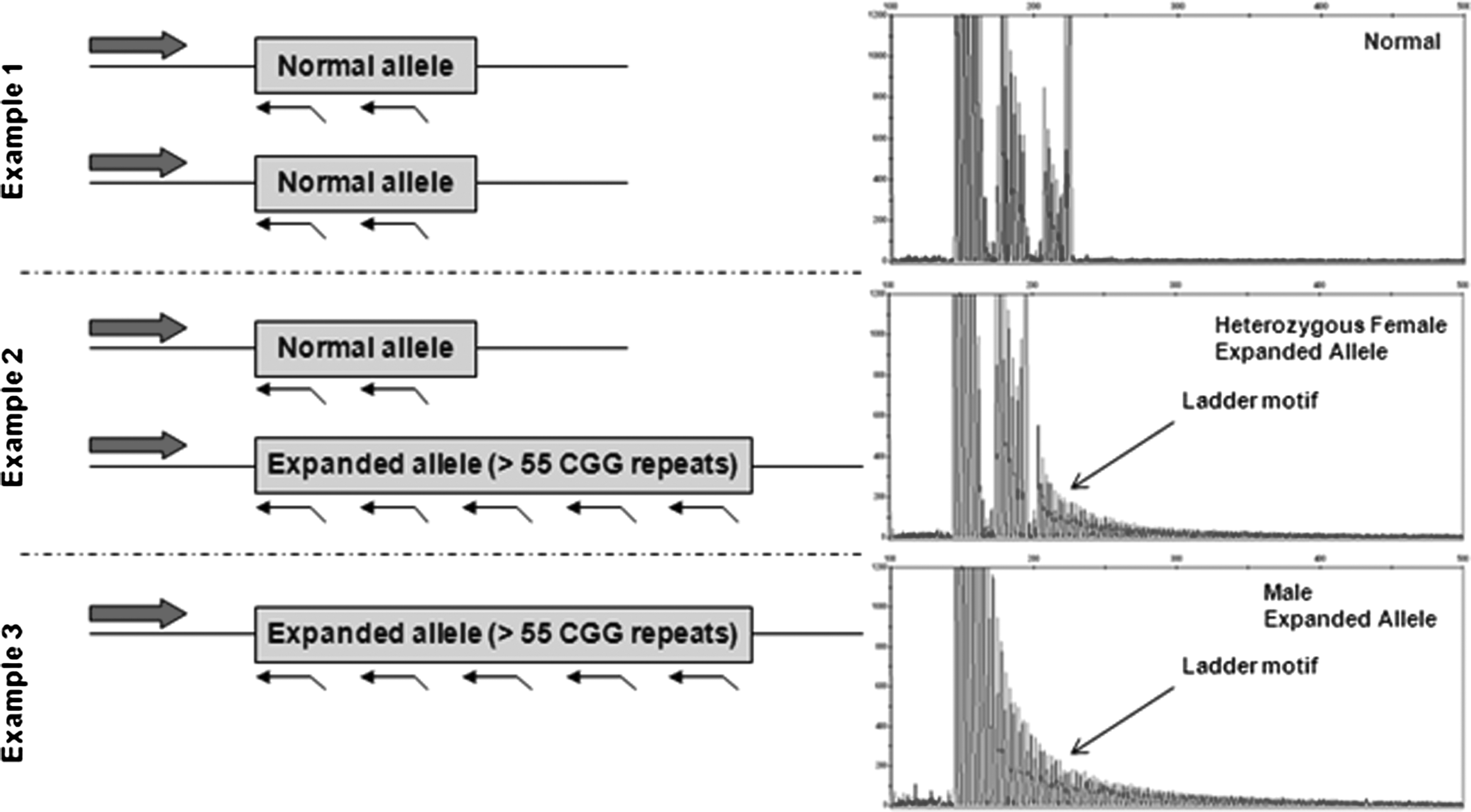
Triplet repeat-primed FMR1 polymerase chain reaction (PCR) assay on Fragile X alleles. A pair of PCR primers generates different sized amplicons depending on the size of the CGG repeat region present. The forward PCR primer is located upstream of the FMR1 CGG region, while the fluorescently labeled reverse primer randomly binds inside the FMR1 CGG repeat region. The presence or absence of a trinucleotide ladder can be easily identified by this assay, and a threshold of 299 base pairs (55 CGG repeats) is typically used to define expanded FMR1 alleles. Example 1 shows the trace for a female with two normal alleles, neither of which results in the characteristic ladder motif present for expanded alleles. Example 2 illustrates the typical pattern for a female with one normal allele and one expanded allele, while Example 3 illustrates the typical pattern for a male with an expansion at the FMR1 locus.
Results
Validation of the laboratory-developed triplet repeat-primed FMR1 PCR assay
Using a threshold of 55 CGG repeats, we identified the presence of expanded FMR1 alleles in 83 of the samples included in this validation study (40 premutation-sized alleles, 37 full mutations, and 6 mosaics). The remaining 629 validation samples yielded normal or intermediate (nonexpanded) results. The six mosaic samples evaluated during the validation phase included four premutation/full mutation mosaics and two full mutation methylation mosaics. The results obtained from the high-throughput lab-developed test using Celera's reagents demonstrated 100% concordance with the previous result obtained during routine diagnostic testing at the Greenwood Genetic Center (Table 1). This yields an analytical sensitivity of 100% (95% confidence interval is 95.58-100) and an analytical specificity of 100% (95% confidence interval is 99.39-100) for the laboratory-developed triplet repeat-primed FMR1 PCR assay (Hemant et al., 2011).
A total of 712 samples were tested during the validation phase of this study; 83 had expanded alleles (11.6%). Original diagnostic test results obtained by the Molecular Diagnostic Laboratory of the Greenwood Genetic Center are shown in addition to the results obtained during the validation of the high-throughput laboratory-developed triplet repeat-primed FMR1 PCR assay. All validation results obtained with the triplet repeat-primed PCR assay were 100% concordant with the previous molecular results obtained by routine diagnostic testing. This demonstrates the power and reliability of the FMR1 PCR assay utilizing Celera's ASR and GPR reagents.
ASR, analyte-specific reagents; GPR, general purpose reagents; PCR, polymerase chain reaction.
To further test the sensitivity of this assay, synthetic female samples were created by combining DNA samples from two males with differing allele sizes. This allowed the opportunity to challenge the assay with potentially difficult allele combinations that are rare in the general population. Briefly, 16 different synthetic female samples were created so that two X chromosomes were represented, each with either a normal or low premutation-sized allele and each with either a premutation-sized allele or full mutation. All synthetic female samples yielded appropriate results for the allele sizes present in each sample (Fig. 2).
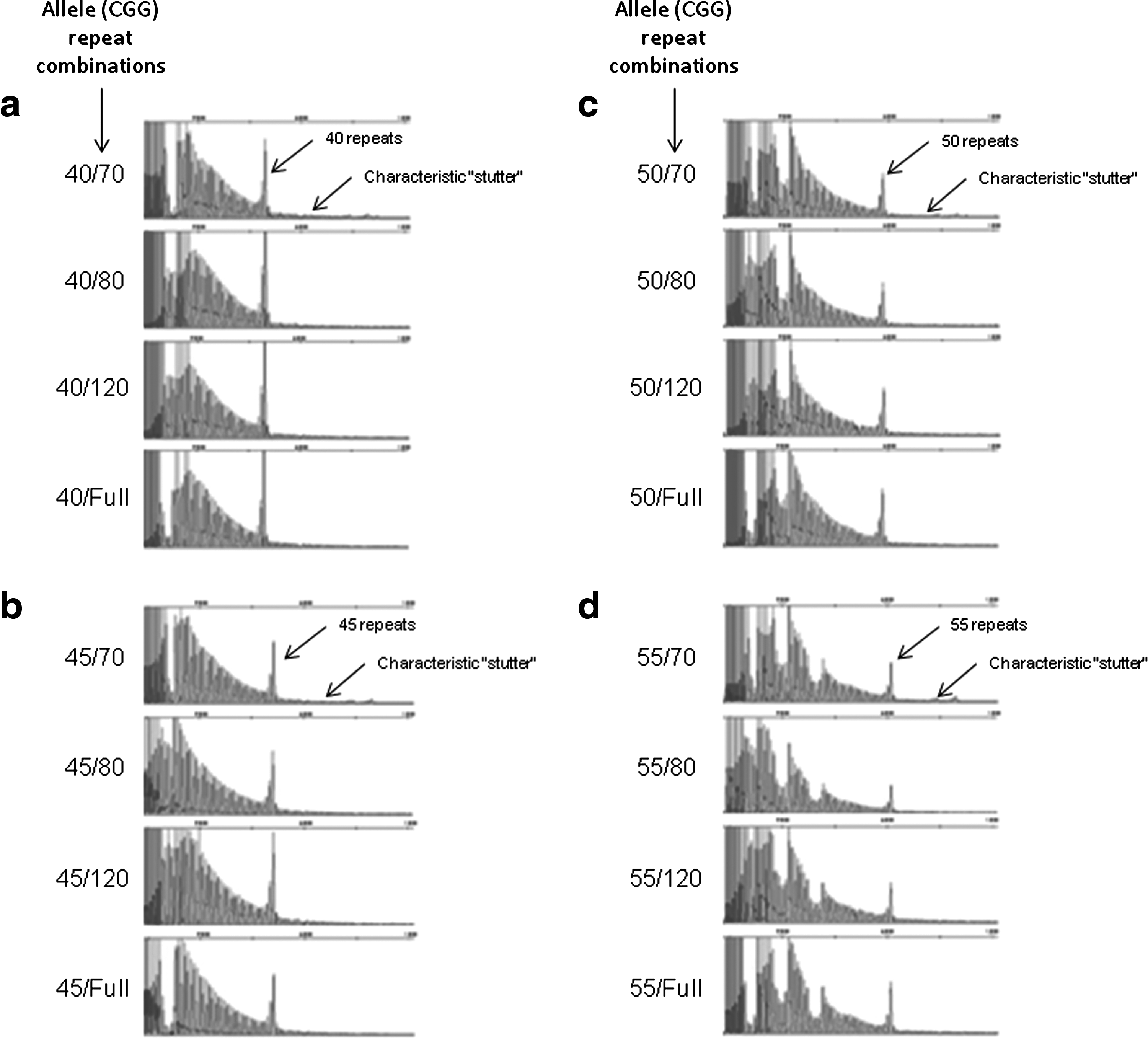
Analysis of synthetic female samples using the triplet repeat-primed FMR1 PCR assay. Sixteen different combinations of synthetic female samples were created by combining DNA samples from two males with differing allele sizes. The allele (CGG repeat) combinations are shown to the left of each panel of data and are as follows:
Additionally, during the course of the validation of the triplet repeat-primed FMR1 PCR assay, reproducibility studies were also performed for the first cohort consisting of 88 samples with a high percentage of expanded alleles. An aliquot of DNA from all 88 samples was sent from the Molecular Diagnostic Laboratory at the Greenwood Genetic Center to the Celera Laboratory for analysis using the laboratory-developed triplet repeat-primed FMR1 PCR assay. Using a cutoff of 55 CGG repeats to differentiate the expanded samples from the nonexpanded samples, there was 100% concordance in results obtained, demonstrating reproducibility in results from the two independent laboratories.
Diagnostic testing with the laboratory-developed triplet repeat-primed FMR1 PCR assay
The current diagnostic testing strategy at the Greenwood Genetic Center requires that all samples be concurrently tested using a Greenwood Genetic Center (GGC)-developed fluorescent PCR assay and the laboratory-developed triplet repeat-primed FMR1 PCR assay utilizing Celera's ASR and GPR reagents. This diagnostic strategy was implemented after validation of the two independent sample cohorts that provided the necessary confidence to move forward in this direction. A conservative approach is taken whereby samples with 45 or more CGG repeats are reflexed for Southern blot analysis (Fig. 3). For the 8 months that this testing strategy has been in place, a total of 437 patients have been submitted to our laboratory for diagnostic testing. Of the 437 samples, only 28 of them (6.4%) required reflex Southern blot analysis. Details, including gender and allele size(s), for all of the samples with 45 or more CGG repeats are reported in Table 2.
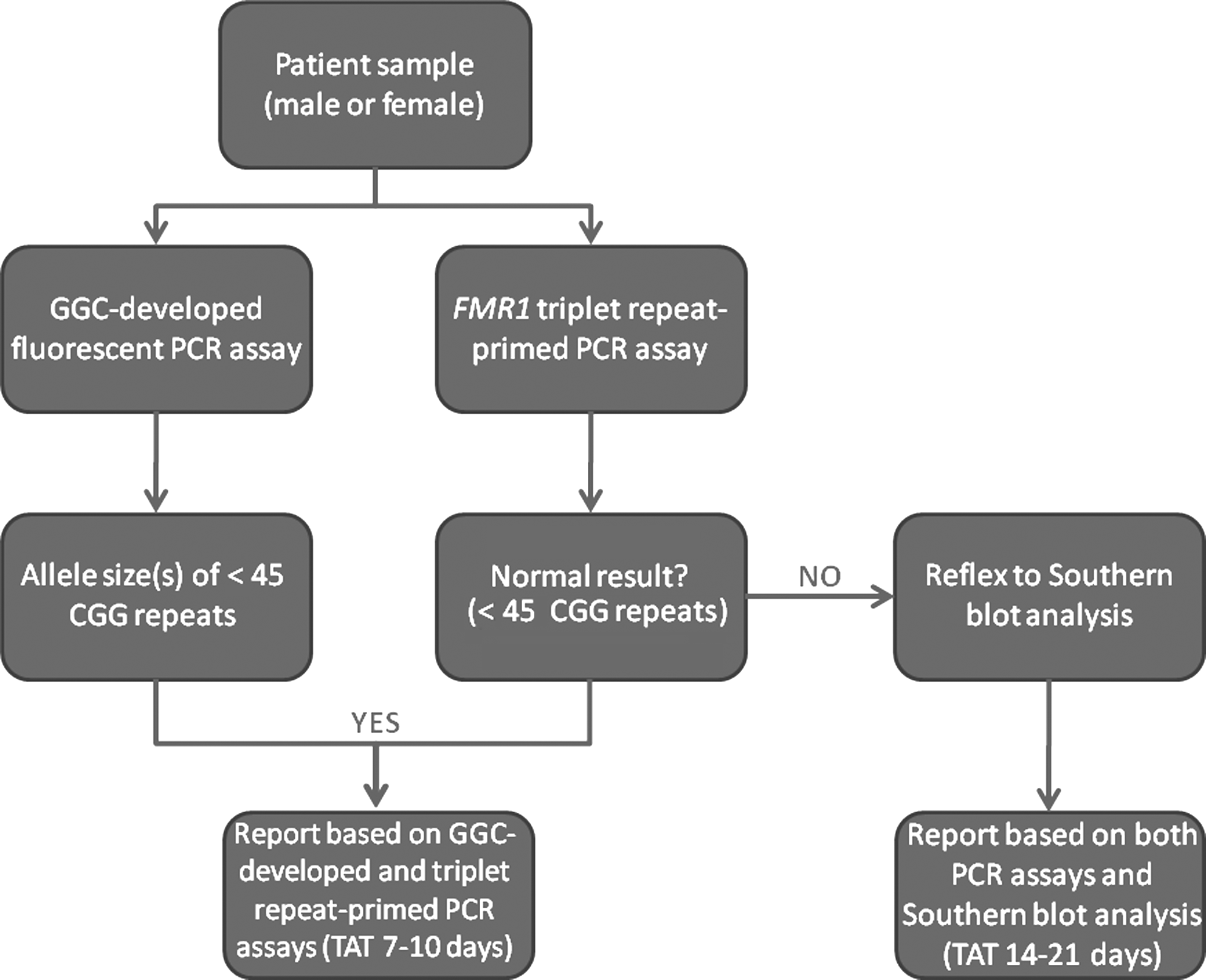
Current diagnostic testing strategy at the Greenwood Genetic Center. The current diagnostic testing strategy at the Greenwood Genetic Center uses a cutoff of 45 CGG repeats. All samples with<45 CGG trinucleotide repeats using the GGC-developed and triplet repeat-primed FMR1 PCR assays are considered normal and are reported based on these PCR assays alone. Samples with≥45 CGG trinucleotide repeats fall into the intermediate, premutation, or full-mutation categories and are reflexed to Southern blot analysis to determine size and methylation status. The turnaround times (TAT) for each scenario are also shown.
A total of 437 patients have been evaluated for Fragile X at the Greenwood Genetic Center using the triplet repeat-primed FMR1 PCR assay. Only 28 samples (6.4%) were reflexed to Southern blot analysis before the reporting of results, 17 of which had≥45 CGG repeats. Details for the 17 samples with≥45 CGG repeats are shown. The remaining 11 samples required Southern blot analysis because they had an atypical pattern by the GGC-developed fluorescent PCR assay (n=9) or a triplet repeat-primed PCR assay failure (n=2). However, these 11 samples were ultimately reported to be normal as subsequent testing indicated that they did not have an expansion at the FMR1 locus.
Discussion
Most individuals with intellectual disability, developmental delay, and/or autism are tested for Fragile X syndrome given the fact that the diagnosis can easily be overlooked, especially in younger patients, and that the condition is relatively common in the world of clinical genetics. For years, diagnostic testing has revolved around using both PCR and Southern blot analysis to analyze the number and methylation status of the FMR1 CGG trinucleotide repeat element. These complementary methods have different levels of resolution and provide slightly different pieces of information regarding the number of CGG repeats. Southern blot analysis is a labor-intensive method that typically takes most laboratories a week to complete. Attempts to move away from the necessity of doing Southern blots have been considered an area of renewed interest in recent years. Tassone et al. (2008) reported the first use of an in-house developed PCR strategy for the rapid identification of expansions in FMR1; however, this assay relied on agarose gels instead of capillary gel electrophoresis. The newer, previously described laboratory-developed triplet repeat-primed FMR1 PCR assay (Lyon et al., 2010) makes use of a fluorescently labeled primer and capillary gel electrophoresis, thereby increasing the ease and certainty with which results can be interpreted.
The current diagnostic testing strategy at the Greenwood Genetic Center couples the laboratory-developed triplet repeat-primed FMR1 PCR assay that allows for the identification of the presence of larger expansion mutations with a standard GGC-developed fluorescent PCR for sizing of normal, intermediate, and smaller premutation-sized alleles. Under this strategy, all samples are concurrently tested using both the GGC-developed and triplet repeat-primed FMR1 PCR assays. When the GGC-developed PCR assay indicates that a sample has<45 CGG repeats and the triplet repeat-primed FMR1 PCR assay does not identify the presence of an expanded allele, testing is considered to be complete and the sample is reported to be normal. The molecular test reports for these samples are generated within 7-10 days from the time the sample is received by our laboratory. However, when samples are interpreted to have ≥ the 45 CGG repeat threshold by both PCR assays, they are reflexed to Southern blot analysis for an estimation of their size and determination of their methylation status. For these samples, the triplet repeat-primed FMR1 PCR data are considered to be supplemental and complement the Southern analysis. Molecular test reports for intermediate, premutation, and full mutation samples that require Southern blot analysis are generated 14-21 days from the time that the sample was received by our laboratory.
This diagnostic testing strategy has been in place for 8 months in our laboratory, during which, a total of 437 patients have been submitted for diagnostic testing. Of the samples tested to date, only 28 (6.4%) required reflex Southern blot analysis. Therefore, the turnaround time for 93.6% of the samples submitted to our laboratory for analysis of Fragile X syndrome was decreased from 14-21 days to 7-10 days. Seventeen of the samples that required Southern blot analysis included six premutation samples, six full mutation samples, four intermediate samples, and one premutation/full mutation mosaic sample. Gender and allele sizes identified for these 17 samples are reported in Table 2. It is important to note that none of the four intermediate-sized alleles had any evidence of abnormal methylation. In addition, one XXY male, one XXX female, and nine normal samples also required Southern blot analysis before reporting out molecular test results. Given the fact that a male sample had two distinct allele sizes by the GGC-developed fluorescent PCR, Southern blot testing was ordered to evaluate the methylation status and confirm the presence of two X chromosomes. This also ruled out any possibility of a sample switch during the GGC-developed fluorescent PCR assay. Similarly, for the female sample with three distinct allele sizes by the GGC-developed fluorescent PCR, Southern blot testing was also ordered; however, subsequent chromosome analysis revealed a 47, XXX karyotype confirming the presence of three X chromosomes. Two of the nine normal samples required reflex Southern blot analysis due to a triplet repeat-primed PCR assay failure, while the remaining seven normal samples required reflex Southern blot analysis due to an unusual pattern observed by the GGC-developed fluorescent PCR assay (most often a pattern which resembled that of two overlapping alleles was observed). For all 11 of these cases, Southern blot analysis resolved the issue at hand and allowed the molecular test results to be reported as normal (no evidence of trinucleotide expansion at the FMR1 locus) with certainty.
This assay validation study further demonstrates the utility of using triplet repeat-primed FMR1 testing in a clinical diagnostic setting. An assay of this type is desirable for both clinical diagnostic laboratories and laboratories offering population-based screening as it quickly identifies the presence or absence of an expansion mutation. When comparing two different 6-month time periods, both before and after integration of the triplet repeat-primed FMR1 PCR assay into our routine diagnostic workflow, we experienced a 75% reduction in the number of Southern blots required when using the triplet repeat-primed FMR1 PCR assay. Since the number of samples requiring Southern blot analysis is significantly reduced, the success of triplet repeat-primed PCR strategies allows the opportunity for a significant decrease in the turnaround time for most samples tested with a cost savings realized with regard to both reagents and time. Although there are many evolving issues that need to be addressed regarding the practicality of population-based screening for newborns and women of reproductive age (Coffee, 2010; Hantash et al., 2010; Hill et al., 2010), our results further support the notion that higher throughput testing for population screening purposes is becoming increasingly possible, both from a cost and a time perspective.
Footnotes
Acknowledgments
The authors would like to thank Celera Corporation and Abbott Molecular for providing materials for the study. We also thank Sara Sarasua and Julianne Collins of the Greenwood Genetic Center for their bioinformatic assistance.
Disclosure Statement
N. Marlowe, T. Laver, and D. Behlendorf are employed by Celera Corporation, manufacturer of the FMR1 analyte-specific and general-purpose reagents.