
Abstract
Since the late 1990s, there has been an unprecedented growth in the development of new molecular and proteomic assays for clinical decision making. Despite the thousands of tests available, a standardized, well-defined, and coherent evaluation framework for these molecular assays is still lacking. We aim to summarize the publicly available appraisal criteria and to develop a succinct and accessible set of criteria that can provide a roadmap for the appraisal of gene-based laboratory developed tests (LDTs). We conducted a systematic literature review of the available molecular diagnostic framework in PubMed MD and CINAHL and identified 91 articles on existing appraisal criteria. We provided a summary of the historical appraisal system and developed an analysis of these appraisal systems, LDT-SynFRAME, which details the major criteria for evaluating molecular diagnostics in the clinical setting. Our goal with the LDT-SynFRAME system is to promote a well-informed dialog among all the stakeholders responsible for the development, approval, reimbursement, and use of new molecular classifiers.
Introduction
T
With the promise of each new assay entering clinical practice comes the question of what processes should be followed to appraise its effectiveness, safety, utility, and affordability. The audience for a standardized set of appraisal processes is large and has diverse backgrounds and interests; it includes regulatory officials, health technology appraisal groups, investors, developers of molecular assays, clinical investigators, payers, physicians, and the general public.
To meet this need, several health technology assessment (HTA) groups have adapted their existing appraisal processes, many of which were developed initially for evaluating pharmaceuticals, for the assessment of molecular assays and LDTs (U.S. Preventive Services Task Force Procedure Manual 2008; Wald and Cuckle, 1989; Hayes et al., 1996; Haddow and Palomaki, 2003; McShane et al., 2005; Simon, 2006; Gene Expression Profiling for Managing Breast Cancer Treatment, 2007; Genetic Testing, 2007; Teutsch et al., 2009). Consensus and standardization of the appropriate terminology, definition of these terminologies, and appropriate evaluation criteria, however, are lacking. This discordance is best highlighted by the inconsistent use of clinical utility and clinical validity by the clinicians, researchers, policy makers, and appraisal groups. In addition, confusion remains on the necessary and sufficient steps required to validate a novel assay, reflected by on-going debate on the appropriate market authorization and regulation oversight required for LDTs (Ntzani and Ioannidis, 2003; Michiels et al., 2005). The primary aims herein are to (1) summarize publicly available appraisal criteria and (2) to develop a succinct and accessible set of criteria that can provide a roadmap for the appraisal of gene-based LDTs.
Methods
We searched PubMed (U.S. National Library of Medicine, Bethesda, MD) and CINAHL (Cumulative Index to Nursing and Allied Health Literature, EBSCO Publishing, Ipswich, MA) from 2000 to the present to identify literature that explains processes used to appraise a molecular assay. We also conducted an ISI Web of Science® (Thomson Reuters, New York, NY) supplemental search of key articles and looked at guidelines that are published by government agencies, professional societies, and HTA groups. Search terms included <molecular><molecular assay*> <molecular test*> <validation> <clinical validation> <analytical validation> <clinical utility> <clinical impact> <cost-eff*> <cost-benefit> <economics>. Inclusion criteria were (1) description of framework(s) or processes for assessing molecular assays, (2) explicit appraisals of molecular assays that included a description of the appraisal process, (3) other guidelines that have been published on appraisals, and (4) English language. We exclude case reports, editorials, and reviews (Fig. 1).

Literature search flow diagram.
Two authors (J.H. and J.D.) reviewed and abstracted all articles to identify the criteria deemed to be important when appraising molecular assays. We organized these criteria into several recognized categories, thereby creating a structured technology appraisal framework for molecular classifiers and gene-based LDT.
Results
The literature search retrieved 1136 articles, of which 24 were duplicates and 1021 articles were excluded. Ninety-one articles were reviewed (Fig. 2). Articles that did not meet the inclusion criteria were excluded from the final summary. In addition, evaluation methods from prominent HTA organizations were retrieved and incorporated into our validation methodology. Brief accounts of the history of the appraisal process, as well as its current state, were drawn from the literature.
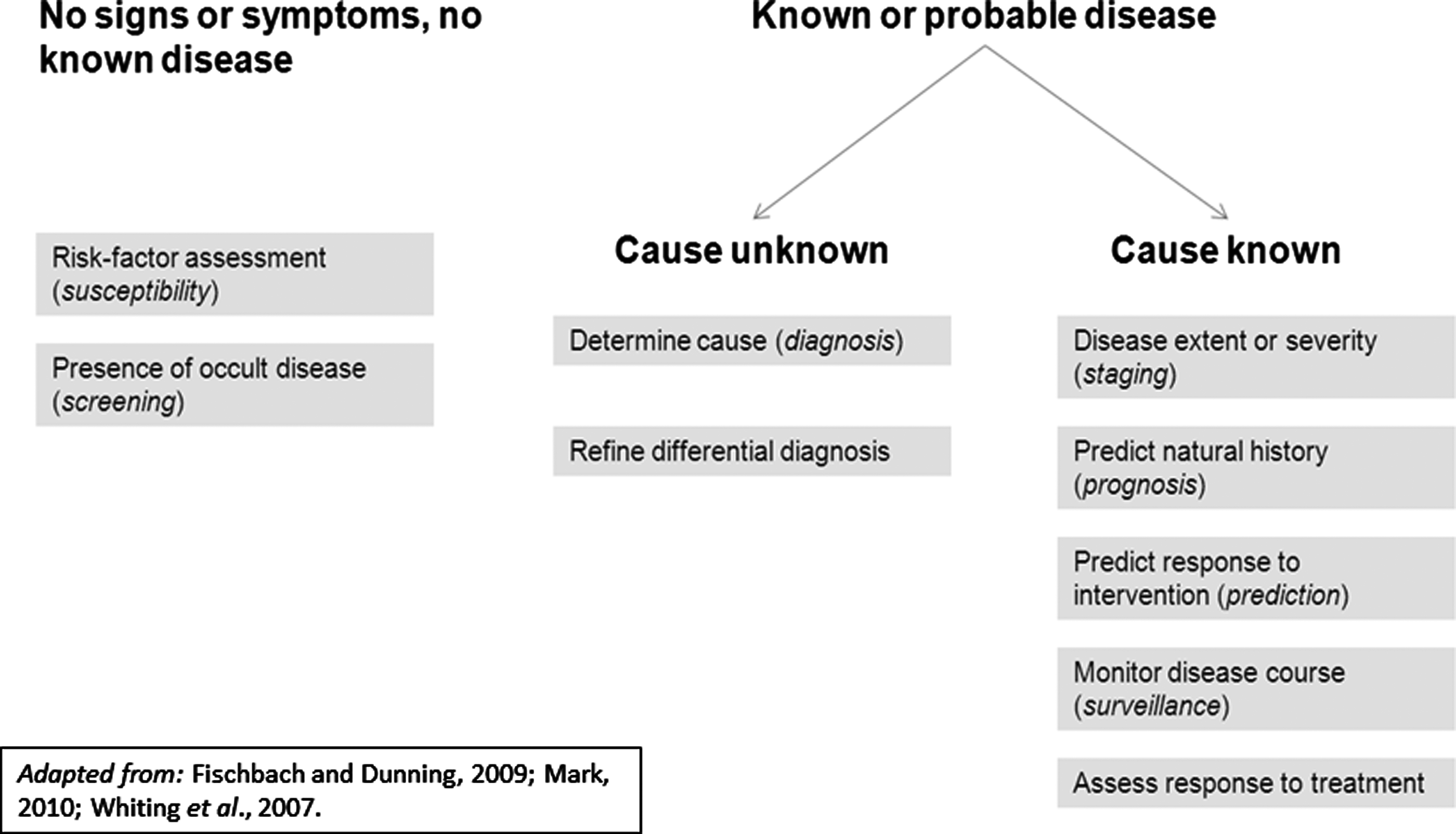
Categories of tests.
Classification of tests
A wide variety of molecular tests are available. Based on canonical medical texts, we created a basic classification system for LDTs shown in Figure 2. Two categories of test were identified: first tests for signs, symptoms, or no known disease; second tests for known disease. Under the first category, there are tests for susceptibility or risk-factor assessment to initiate intervention and prevent occurrence of disease, such as the BRCA1 gene test for risk of breast cancer (Weitzel et al., 2007). There are also tests for screening the presence of occult disease, to initiate intervention to cure or avoid progression to more severe health states, such as the Papanicoloau test for diagnosis of precancerous or cancerous cervical lesions (Weitzel et al., 2007).
Under the category of tests that pertain to apparent disease, there are tests that diagnose or determine cause of apparent symptoms to decide on interventions, for instance, chest X-ray for a patient with chronic cough (Hricak et al., 2005). Another type of test develops or refines a differential diagnosis, to reduce the list of possible causes of prior clinical or test findings, such as an electrocardiogram for abnormal pulse (Pearson et al., 2002). Tests for the management of known diseases consists of five categories. Staging tests that evaluate the extent or severity of the disease, as well as prognostic tests that predict natural history, assess the urgency of the problem, and the appropriateness of intervention. Tests that predict response to treatment help to inform selection of an intervention, such as the 21-gene recurrence score for early-stage breast cancer (Paik et al., 2006). Another type of test monitors the course of the disease to assess the disease status and the need for intervention. Lastly, tests that assess response to intervention are meant to assess the effectiveness of treatment. Table 1 provides examples of these categories of tests. We aim to describe a unified validation framework for the wide diversity of the aforementioned tests.
β-HCG, beta-human chorionic gonadotropin; CEA, carcinoembryonic antigen; CT, computed tomography; EKG, electrocardiogram; GCC, guanylyl cyclase C; MRI, magnetic resonance imaging; Pap, Papanicoloau.
Appraisal process: a brief history
Table 2 describes the major efforts to create appraisal systems over the last 20 years. These efforts include the Tumor Marker Utility Grading System (TMUGS), the U.S. Preventive Services Task Force, Blue Cross Blue Shield Technology Evaluation Center (BCBS TEC), the Centers for Disease Control and Prevention Analytical Validity, Clinical Validity, Clinical Utility, Ethics, Legal, Social Implications system, (CDC ACCE) REporting Recommendations for MARKer Prognostic Studies (REMARK), and others (Wald and Cuckle, 1989; Frame and Carlson, 1975; Hayes et al., 1996; Harris et al., 2001; Haddow and Palomaki, 2003; McShane et al., 2005; Ramsey et al., 2006; Teutsch et al., 2009). These appraisal systems have introduced themes in the validation process that are common to the most widely used appraisal systems currently in use. In general, three major categories of appraisal are cited: analytical validity, clinical validity, and clinical utility. Financial, ethical, societal, and legal implications represent the fourth category that is not consistently included by all assessment agencies, with the evaluation criteria by BCBS TEC as one example. These categories are originally featured in the current framework of the CDC ACCE framework, a commonly cited evaluation method in genetic testing (Genetic Testing, 2007; Teutsch et al., 2009). Analytical validity is defined as the ability of the test platforms, such as microarray or polymerase chain reaction to accurately and reliably measure the analyte of interest. Clinical validity is most often defined as the test's ability to detect or predict the associated disorder. Clinical utility, unlike analytical validity, has a variety of definitions. In a 2008 report, the Johns Hopkins University Evidence-based Practice Center, part of Agency for Healthcare Research and Quality (AHRQ), defined utility as “the clinical utility of a test tells us whether the test helps discriminate between those who will have more or less benefit from a therapeutic intervention” (Marchionni et al., 2008). More recently, AHRQ described clinical utility with a broader definition of “the usefulness of test and its value to clinical practice” (Sun et al., 2010). The Academy of Managed Care Pharmacy defines clinical utility as the safety and efficacy profile of a test (The AMCP format, 2010). In addition to the four ACCE criteria, other authoritative sources stress the importance of other themes, such as appropriate introduction of the disease and test, scientific rigor and validity, clarity in presentation of findings, a rigorous chain of evidence, generalizability, and clear, and unbiased presentation of the evidence (U.S. Preventive Services Task Force Procedure Manual 2008; Hayes et al., 1996; McShane et al., 2005; Simon, 2006; Teutsch et al., 2009). Figure 3 displays a timeline of the seminal appraisal systems over the past three decades. As an analysis of these appraisal systems, LDT-SynFRAME includes the necessary components relevant to the validation of health technology. These components are as follows: a clear introductory context; well-substantiated analytical validity, clinical validity, and clinical utility; economic and ethical outcomes; and transparent presentation. HTA groups have articulated this important factor, such as the U.K. National Institute for Health and Clinical Excellence (NICE) in its scoping process. We emphasize the importance of the evidence being presented in a clear, unbiased, and understandable manner. Third, this is the first published version of the framework, but we anticipate that appraisal processes will continue to evolve, expecting that this continue to evolve across a continuum of time and expert input in a variety of fields.
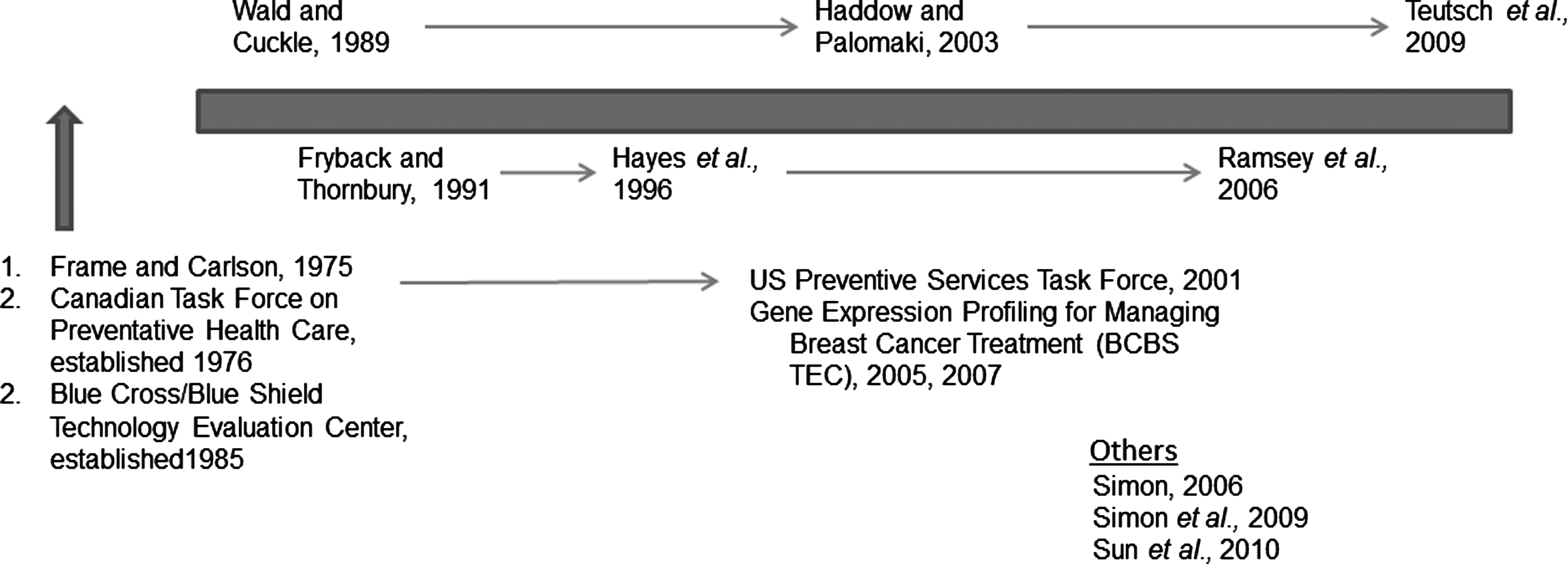
Timeline of key appraisal proposals.
Contextualizing the problem: clear introduction
LDT-SynFRAME combines the most pertinent evaluation criteria from the current leading assessment systems to create one framework with wide applicability to novel tests. The detailed framework is presented in Tables 3-6. At the beginning of the evaluation should be an Introduction, which adequately describes the purpose of the test, the natural history, prevalence and current management of the disorders of interest, and the expected clinical, economic, and social outcomes. The introduction should provide a clear picture of the unmet clinical needs, as well as set a structured context for the application of a novel test. A clear, succinct, and easily read introduction informs an outside reader of the clinical, social, and or economic parameters of the test. Following the introduction are the four major evaluation categories, which are described below.
+, test characteristics relevant to assessing this type of test; NA, test characteristic not applicable to this type of test; x, indicates that definition varies by type of test; y, various academies, colleges, and institutes (see below) still evolving toward convergent standards; blank cells, neither varies by type of test nor is there a relevant different across tests on standards for this test characteristic.
Compiled from standards published by National Academy of Clinical Biochemistry (NACB), the College of American Pathologists (CAP), the Association for Molecular Pathology (AMP), the Clinical Laboratory Standards Institute (CLSI), and the National Institute for Standards and Technology (NIST).
Analytical validity
Mansfield et al. define analytical validation “as a process by which the measurement performance of a test system is assessed (Mansfield et al., 2005).” Traditionally, components of analytical validity have included accuracy, sensitivity, specificity, efficiency, linearity, precision, quality control, traceability, assay stability, sample stability, detection limit, expected values, normalization, success rate, and clear assay cut-off values. The list of criteria was compiled from the standards of the National Academy of Clinical Biochemistry (NACB), the College of American Pathologists (CAP), the Association for Molecular Pathology (AMP), the Clinical Laboratory Standards Institute (CLSI), and the National Institute for Standards and Technology (NIST) (Tholen et al., 2003; Tholen et al., 2004; Mansfield et al., 2005; Krouwer et al., 2006; Wolff et al., 2007; College of American Pathologists, 2007). How these criteria are applied in genotyping, gene expression, and protein expression tests is shown in Table 4 and illustrates how certain elements have yet to reach convergence among different professional societies. Successfully reaching convergence among this diverse range of professional groups on defining appropriate criteria and methods for measuring diagnostic test performance will require overcoming a number of barriers. The high complexity test types enabled by new genomic and proteomic technologies do not fit seamlessly into legacy templates of analytical validation. LDT-SynFRAME is the first framework that pinpoints important distinctions when validating single-gene assays, multi-gene expression assays, and protein expression assays. Traditional measures of diagnostic test performance will have to evolve to adapt to these new, high complexity test types. Furthermore, unique aspects of genetic, genomic, and proteomic tests may mean that developing a single check list of performance criteria to accommodate all test types may not be possible.
Clinical validity
Clinical validity refers to the extent to which a test accurately predicts the risk of an outcome (i.e., calibration), as well as its ability to separate patients with different outcomes into separate risk classes (discrimination) (Marchionni et al., 2008). In the case of clinical decision making for early stage breast cancer, clinical validity refers to ability of the test to (1) predict recurrence risk or (2) treatment response.
Several research groups have commented on processes they use to evaluate the clinical validity of molecular classifiers (Gene Expression Profiling for Managing Breast Cancer Treatment, 2005; Simon, 2006; Marchionni et al., 2008). Although these groups address specific aspects of clinical validity, we found that they differ in their specific definitions and in how criteria are organized. We grouped the criteria for clinical validity into four categories: design, sample population, clinical meaningfulness, and statistical significance.
For a sample population, experts recommend that clinical validation studies enroll patients who are representative of the target population. Sufficient estimation of treatment benefit is accomplished in the context of randomized controlled trials. This may be done prospectively or retrospectively, given that all measurements are done concurrently and independently of the outcome. If results of retrospective analyses of randomized clinical trials are consistent in two or more independent studies, then the evidential value of the retrospective study is comparable to that of a concurrent prospective study (Simon et al., 2009).
Simon recommends that the patients' characteristics should be sufficiently homogeneous to assure that they face similar therapeutic options (Simon, 2006). For example, the treatment options for women with node-positive breast cancer are sufficiently different from treatment for women with node-negative disease. A study that includes both stages of cancer would be insufficiently homogeneous to draw meaningful conclusions about the validity (and utility) of the test in real-world settings. Patients should be enrolled in therapeutically meaningful clinical studies to assess if the classifier is valid as a predictor of not only prognosis but also treatment response.
Last, the study must be large enough to draw meaningful statistical inferences and assess generalizability to other populations. A suggested rule of thumb is that the study provides at least 20 patients per class (e.g., 20 responders and 20 nonresponders) (Simon, 2006).
Clinical meaningfulness criteria pertain to validation studies properly examining endpoints that influence decisions in the clinic. Studies should examine endpoints that are clinically relevant; in the case of adjuvant chemotherapy for breast cancer, the endpoints should include those typically assessed in clinical trials of pharmaceuticals, such as progression-free survival and overall survival. Data on the endpoints should be obtained rigorously (e.g., limited missing data) and accurately (e.g., limited measurement error). The Johns Hopkins University Evidence-based Practice Center highlighted the importance of cutoffs being stated and pre-specified for the classifier (e.g., low, intermediate, or high) to help determine decisions based on the test result in the 2008 report on the values of the gene expression assay for breast cancer (Marchionni et al., 2008). Last, the validation study should address whether knowledge of the results has clinical implications.
Clinical utility
Clinical utility refers to the ability of a novel tool to add value and clinical benefit beyond traditional or previously established practices. The BCBS TEC defines this criterion as “a technology that should improve net health outcomes as much as, or more than, established alternatives” (Technology Evaluation Center Criteria, 2008). A tool with meaningful clinical utility should lead to a more favorable outcome than the leading standard of care or a suitable comparator. Furthermore, a clinically useful assay should have substantial and measurable impact on clinical decisions. Sparano and Solin in 2010 identified four measures that quantify and qualify the impact of these tests (Sparano and Solin, 2010):
1. Treatment-sparing: test results indicate that treatment will not be beneficial and patient may be spared. 2. Treatment selection: test results indicate that treatment is likely to be beneficial when clinical features indicate no treatment is necessary. 3. Treatment direction: test results provide definitive treatment direction when clinical features are uncertain 4. Treatment confirmation: test confirms original treatment recommendation.
For example, the value of a predictive tool is to reduce uncertainty, which then alters and/or improves clinical decisions and, finally, results in improved clinical outcomes and/or lower costs. The types of uncertainty targeted by an assay may include susceptibility to disease, probability of a diagnosis or set of diagnoses, severity of disease, risk of disease recurrence, risk of toxicity to therapy, and probability of response to treatment(s).
Besides evidence on the assay's ability to influence clinical decisions and outcomes, HTA groups seek evidence on the generalizability of the assay outside of the research settings. The benefits of the assay must reach the general population at large, in that decision makers, such as physicians and patients, will respond practically to the assay's results, and that use of the assay is associated with favorable outcomes and/or lower costs. Some of these may be hard to establish at the time an assay becomes available. For example, how generalizable an assay is and whether it influences decision making may be relatively uncertain until it has been used outside a research protocol.
Financial, social, legal, and economic implications
HTA groups also have been concerned about a broader set of issues pertaining to the adoption of new technologies, which are relevant to molecular assays. These considerations include the financial implications to different stakeholders (e.g., payers, patients, and society as a whole), the relative tradeoffs between financial costs and clinical benefits, how the new assay may differentially affect different populations (especially if it may create widening of disparities in health care), and nonmedical implications, such as access to life insurance and employment.
Economic validity refers to the completeness, quality, and reliability of the analyses used to assess the economic implications of novel technologies. In 2003, Weinstein et al. outlined the criteria that should be considered when evaluating economic analysis; these are detailed in Table 5 (Weinstein et al., 2003). With regard to structure, the objective of the economic analysis must be clearly stated, as well as the parameters and assumptions of the analysis. Data sources must be transparent, graded for quality, and obtained in a well-established methodical way. Analysis of the data should be consistent with well-established statistical methods, and have appropriate patient subgroups analyzed. The instability or uncertainty of the model under varying conditions must be tested. Last, the analysis should have internal consistency, face validity, be calibrated, and subjected to peer review.
Presentation
Presentation is the final component of LDT-SynFRAME and emphasizes the importance of transparent and informative communication of scientific and research methods that is complete, uniform, unbiased, and understandable. This issue was highlighted more than 30 years ago in a 1979 study conducted by Casscells et al. (1978), which showed that “almost all physicians confused the sensitivity of a test with positive predictive value.” In a review of the Casscells et al. (1978) study, Hoffrage et al. (2000) conclude that representation of evidence is critical to understanding and decision making. Evidence, especially statistical data, to support the validity and utility of a test can be conveyed in a number of methods. The accuracy of a diagnostic test could be represented through contingency tables, in numerical values for sensitivity and specificity, or graphically by area under the curves, boxplots, likelihood ratio nomograms, or by decision trees (Whiting et al., 2008). As another example, test prediction can also be presented through a variety of graphs: Kaplan-Meier curves of different cohorts are one possibility, a continuous risk curve is another possibility, and categorical group is a third. While these are all valid methods to present data, not all options are as equally accessible or comprehensible. Hoffrage et al. (2000) recommended that statistical evidence and data must be presented in a manner that is meaningful for the main users. In the field of molecular classifiers, the end users are often physicians and patients; given such an audience, the evidence for the data and test results should be presented in a manner with clinical significances that can be easily understood.36
Discussion
The development of molecular assays has opened new possibilities for reducing clinical uncertainty, improving patient outcomes, and lowering costs. The clinical values of a number of molecular classifiers have been recognized by clinical associations, such as National Comprehensive Cancer Network (NCCN) and American Society of Clinical Oncology (ASCO) and are incorporated into their respective guidelines. The rapidly expanding field of molecular medicine and genomic-based practice, however, is not like the traditional diagnostic model; as such the old appraisal system for pharmaceuticals does not translate into a sufficient framework for appraisal of molecular classifiers. The growth of genomic medicine calls for a new evaluation and assessment system that is accessible to health technology assessor, managed-care payers, physicians, and patients alike. In response to this unmet need, we have summarized and synthesize the published appraisal criteria into the LDT-SynFRAME framework, which is a comprehensive technology evaluation system.
LDT-SynFRAME synthesizes into one the components of all available frameworks, and several of the most important differentiating aspects bear highlighting. First, some of the prior frameworks refer to tests in general, and so provide relative limited specifics on issues concerning the validation of molecular classifiers. Others have focused on validation of single-gene tests, presumably because the majority of novel tests are of this type. However, there are important distinctions, especially with respect to analytical validity, for tests that contain an array of genes, or genes used in combination with other predictors, including clinical factors or protein-based assays. LDT-SynFRAME makes this evolution transparent. Second, the definition of Level I evidence for clinical validation studies has evolved, even since publication of prior frameworks. LDT-SynFRAME incorporates these new concepts. Third, clear communication and reporting of results has been identified as essential for other areas of technology assessment, such as reporting of methods and results of randomized controlled trials and of economic analyses. By synthesizing prior work, some of which was not specifically focused on genetic tests, LDT-SynFRAME uniquely and properly provides a framework for how to evaluate if the reporting of methods and results is comprehensive and unbiased.
We found substantial convergence across prior frameworks for some areas, which were reflected in LDT-SynFRAME. For example, all approaches indicate the importance of a clear introductory statement of the purpose of the assessment. Moreover, they all comment on the importance of assessing clinical validity of the assays and the need to examine the broader ethical, legal, and social issues concerning adoption of such assays. LDT-SynFRAME deliberately differs from other frameworks on the emphasis its places on the importance of economic analyses than others; this reflects that the authors were unconstrained by potential conflicts with policy concerns that continue into this decade to discourage the explicit use of such analyses when appraising technologies.
Will the adoption of LDT-SynFRAME eliminate the confusion many audiences have when seeing contradictory conclusions about the value of a novel diagnostic? We believe not. The various appraisal groups operate under different incentives, so it should be unsurprising that circumstances arise where they arrive at different conclusions when evaluating the same evidence. One of the important values of any appraisal framework is to provide an explicit and transparent set of principles whereby the conclusions of decision makers can be examined thoughtfully, and whereby decision makers can fully and systematically substantiate the conclusions from appropriate application of the framework. Developers of new molecular classifiers and their investors will more accurately determine what evidence will be needed to warrant positive assessments of their innovations. We believe that LDT-SynFRAME, while comprehensive, is also simple enough that it provides a structured perspective for those interested persons, who though they may not perform the actual appraisal, to assess the thoroughness and validity of the appraisals.
Continued and regular update of a framework for assessing molecular classifiers will be needed. Development of and indications for molecular classifiers are changing rapidly; such robust research activity usually brings attention to bear on new issues unforeseen by prior frameworks. In this case, examples include the developments in analytical validity of multi-gene assays versus single-gene assays and definitions of what constitutes Level I evidence for clinical validity. Areas appearing to deserve more discussion and consensus among experts in the field of assessing molecular classifiers include (1) the role of randomized clinical trials, (2) clarifying the principles for the design of decision impact studies, and (3) best principles for reporting methods and results of clinical validation studies. Our contribution is intended to promote a well-informed dialog among innovators, health technology appraisers, physicians, patients, and those responsible for approval and reimbursement of new molecular classifiers, resulting in the highest quality of clinical and policy decision making.
Footnotes
Disclosure Statement
The author received no funding for the development or preparation of this article. All authors, however, were employees of Cedar Associates LLC at the time of writing.