
Abstract
Mutations in mitochondrial DNA (mtDNA) are one of the most important causes of sensorineural hearing loss, especially in the MT-RNR1 gene. In the present study we have performed mutational screening for m.1555A>G and a region of the MT-RNR1 gene in 303 unrelated patients (including family members of 25 probands) with nonsyndromic hearing loss and 200 controls. Three homoplasmic variants, namely, m.1453A>G, 1462G>A, and 1508C>T, were identified in addition to the known deafness-associated m.1555A>G mutation in the MT-RNR1 gene. All the variants were detected only in the patients and not in the controls. m.1555A>G was detected in three probands amounting to 1.0%. Prediction of RNA secondary structure showed changes in all the three variants, the most severe being in m.1453A>G that was inherited in a typical maternal pattern in two families. Screening of GJB2 and GJB6 genes in all these probands revealed cosegregation of the p.W24X mutation (GJB2) in one family with m.1453A>G. Only the proband carrying the p.W24X mutation in a homozygous state expressed the condition while heterozygous and normal homozygous relatives had normal hearing in spite of having the mutation in MT-RNR1. The conservation index (CI) of m.1453A>G was found to be 82%, suggesting it to be a possibly deleterious mutation. Functional studies using cell lines derived from muscle tissue of these patients may reveal the pathogenic mechanism of deafness in them.
Introduction
O
Sequence analysis of the mitochondrial genome in families and sporadic cases from various ethnic origins provides strong evidence that the MT-RNR1 gene is the hot spot for NSHL in addition to causing genetically determined susceptibility to aminoglycoside ototoxicity. The most common mutations in the MT-RNR1 gene causing NSHL include m.872A>G, m.961delT+insc, m.961T>C, m.1095T>C, m.1494C>T, and m.1555A>G (Prezant et al., 1993; Yoshida et al., 2002; Zhao et al., 2004; Li et al., 2005; Xing et al., 2006). Of these mutations, m.1494 C>T and m.1555A>G substitutions show clear contribution to nonsyndromic and aminoglycoside-induced hearing impairment (Prezant et al., 1993; Guan et al., 2000; Zhao et al., 2005; Kokotas et al., 2009; Zhu et al., 2009; Nahili et al., 2010; Rydzanicz et al., 2010). Other sequence variants are still considered as provisional and their pathogenic role remains controversial, mainly due to their variable frequency in different populations, mtDNA phylogeny, and their occurrence in normal hearing individuals.
Recent studies indicate that the phenotypic expression of mtDNA mutations, especially m.1555A>G, is highly variable, indicating that these mutations do not contribute to the clinical outcome in toto (Guan, 2004; Guan et al., 2006). Some differences in either the nuclear gene content or in activity appear to significantly contribute to the biochemical defect affecting the penetrance of nonsyndromic deafness. Thus, nuclear modifier genes or other modifier factors may modulate the phenotypic manifestation of the MT-RNR1 mutations by interacting with the gene, either suppressing or enhancing the effect of the mutation (Bronya et al., 2006).
Up to date, little is known about the incidence of MT-RNR1 mutations in Indian patients. Hence, in the present study, we made an attempt to screen for the m.1555A>G mutation and other sequence changes in the MT-RNR1 gene in addition to mutations in GJB2 and GJB6 genes in patients with congenital, profound, nonsyndromic hearing impairment.
Materials and Methods
Patients
A total of 303 hearing impaired patients, including family members of 25 probands, cooperated to give blood samples and their DNA was tested for the presence of mutations in MT-RNR1 (mitochondrial 12SrRNA), GJB2, and GJB6 genes. These cases were recruited from the Government Ear, Nose and Throat (ENT) Hospital, Hyderabad, and schools for hearing impaired in and around the districts of Hyderabad. All the probands were examined by the ENT specialists and the loss of hearing was graded based on pure tone audiometry. The inclusion criterion for this study was deafness graded as profound sensorineural hearing loss (≥90 dB). Patients with a history of acquired etiology, genetic syndrome, otoacoustic trauma, or any neonatal diseases were excluded from the study. None of the cases described revealed exposure of their mothers to drugs/teratogens or infections when they carried the probands. Simultaneously 200 healthy controls with normal hearing and without the history of deafness in their family were selected from the same population at random to compare with the data generated on the probands. Pedigrees with a history of four to five generations were constructed for both patients and control individuals.
Individuals were enrolled for the study with their consent after explaining to them the purpose and possible outcome of the investigation. The study was approved by the Institutional Ethical Committee.
Mutation analysis
DNA was isolated from venous blood collected from the probands, family members of 25 probands, and controls using a rapid nonenzymatic method (Lahiri and Nurnberger, 1991).
Screening of the mitochondrial m.1555A>G mutation
Samples were screened for the m.1555A>G mutation by polymerase chain reaction (PCR) amplification of mtDNA, using 50 ng of DNA and 10 pmol of each primer (5′ GCT CAG CCT ATA TAC CGC CAT CTT CAG CAA 3′ [Forward] and 5′ TTT CCA GTA CAC TTA CCA TGT TAC GAC
Single strand conformation polymorphism analysis of the MT-RNR1 gene
For the detection of other mutations in the MT-RNR1 gene, single strand conformation polymorphism (SSCP) analysis was performed for the region (1237-1777) of the MT-RNR1 gene amplified using the primers forward 5′ ACCACCTCTTGCTCAGCCTA 3′ and reverse 5′ CTATTGCGCCAGGTTTCAAT 3′ in a total reaction volume of 10 μL containing 50 ng DNA, 2.5 pmol of each primer, 200 μM of each dNTPs, and 0.25 U Taq DNA polymerase. The conditions for PCR were initial denaturation at 95°C for 5 min followed by 35 cycles of denaturation at 95°C for 45 s, annealing at 55°C for 1 min, extension at 72°C for 45 s, and a final extension at 72°C for 5 min. The PCR products were denatured in low ionic strength buffer (0.01% bromophenol blue, 0.01% xylene cyanol, and 10% sucrose) at 95°C for 10 min and electrophoresed in 12% polyacrylamide gel (37.5:1) with glycerol for≈16 h at 100 V. The gels were visualized by silver staining. All the variants were confirmed by sequencing on ABI 3100 DNA sequence analyzer and the sequence data were compared with the revised Cambridge Sequence, GeneBank accession No. NC_012920 (http://ncbi.nlm.nih.gov/GenBank/).
The nature of all identified 12S rRNA nucleotide changes (polymorphism, putative pathogenic variant, and mutation) was checked by searching the human mitochondrial genome database MITOMAP, mtDB (http://mitomap.org).
Mutational screening of the GJB2 and GJB6 mutations
In addition to the screening for mutations in MT-RNR1 gene, screening for already reported common mutations in Exon1 and Exon2 of GJB2 gene (c. IVS1+1G>A [exon1], p.W24X [c.71G>A], c.35delG, c.167delT, p.W77X [c.231G>A], and c.235delC) was performed using allele-specific PCR amplification (ARMS PCR) and restriction fragment length polymorphism methods and for other novel mutations by SSCP analysis (Padma et al., 2009).
GJB6 gene was screened for del (GJB6-D13S1830) using a specific PCR assay described by del Castillo et al. (2002). Also, SSCP analysis was performed for four overlapping regions of Exon3 of GJB6 gene (Padma et al., 2009).
Secondary structure prediction
The RNAfold software from Vienna RNA package (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) was used to generate and predict RNA secondary structure based on minimum energy requirements and pair probability. The pattern of folding in mutated sequences was compared with the wild-type prediction.
Phylogenetic analysis
A total of 22 primate mitochondrial MT-RNR1 sequences were used in the interspecific analysis. These include Homo sapiens, Gorilla gorilla, Pan paniscus, Pan troglodytes, Pongo pygmaeus, Pongo abelii, Hylobytes lar, Macaca sylvanus, Papio hamadryas, Cebus albifrons, Tarsius bancanus, Nycticebus coucang, Lemur catta, Bos taurus, Mus musculus, Rattus norvegicus, Equus caballus, Balaenoptera physalus, Balaenoptera musculus, Phoca vitulina, Halichoerus grypus, and Didelphis virginiana (Genbank). The conservation index (CI) was calculated by comparing the human nucleotide variants with the other 21 sequences. The CI was defined as the percentage of species from the list of 22 different species that have the wild-type nucleotide at that position.
Results
m.1555A>G mutation
Screening of 303 hearing impaired subjects (including family members of 25 probands) for m.1555A>G transition in MT-RNR1 gene revealed three sporadic cases positive for the mutation and none in the controls. In all the three cases, restriction digestion analysis and subsequent electrophoresis indicated that the mutation was homoplasmic. Audiological evaluations revealed that all the three probands had severe hearing impairment (100 dB). Further clinical data revealed that they had no history of exposure to aminoglycosides and maternal exposure to toxic substances or any infection during pregnancy. The frequency of m.1555A>G thus works out to be 1.0% for Indian patients that is comparable with previous reports on other ethnic groups where the frequency of this mutation ranges from 0.6% to 2.5% (Usami et al., 2000; Hutchin et al., 2001; Kupka et al., 2002; Tekin et al., 2003; Li et al., 2004; Jacobs et al., 2005).
Novel nucleotide changes detected in the MT-RNR1 gene
SSCP analysis of a region (1237-1777) of the MT-RNR1 gene in 303 probands revealed three different patterns of mobility shifts that on sequencing corresponded to three novel variations in the homoplasmic state. They were m.1462G>A, m.1508C>T, and m.1453A>G, which were submitted to the Human Mitochondrial Genome Database (http://mitomap.org) with the reference numbers #20081106002, #20081106001, and #20081105001, respectively.
The m.1462G>A variant was detected in two unrelated probands with severe hearing impairment (100 dB) and positive family history. Both the probands had no history of exposure to aminoglycoside antibiotics. Family members of these two probands could not be studied as they were not willing to participate in the investigations.
The m.1508C>T variation was found in one proband with profound hearing loss (90 dB). Further evaluation revealed that he had no exposure to aminoglycoside antibiotics.
The m.1453A>G variant was found in two unrelated probands affected with nonsyndromic hearing impairment, wherein one showed positive family history (mt-01; Fig. 1) and the other was sporadic (mt-02). The analysis of other members of the families of the two probands confirmed the presence of m.1453A>G variation and its inheritance in a typical maternal pattern. In mt-01, the variation was detected in proband's sister (IV-1), mother (III-4), grand mother (II-2), maternal aunt (III-7), maternal aunt's daughter (IV-4), and maternal uncle (III-9) all of whom had normal hearing. Screening of this family for GJB2 and GJB6 mutations revealed the cosegregation of the p.W24X mutation of the GJB2 gene in the proband (IV-2) who was homozygous for the mutation. The proband's father (III-3), mother (III-4), grand mother (II-2), paternal uncle (III-1), maternal aunt (III-7), and maternal uncle (III-9) were heterozygous for the p.W24X mutation but had normal hearing. In mt-02, the proband's brother, mother, grand mother, two maternal aunts, and maternal aunt's son showed the m.1453A>G variation but had normal phenotype. Mutations in GJB2 and GJB6 genes were not detected in this family nor in the proband. The variability in the expression of this mutation in the two families is similar to the findings reported in other nonsyndromic associated mtDNA mutations, such as m.1555A>G or m.7443A>G, which are considered ineffective in producing the clinical phenotype, that is, hearing impairment.
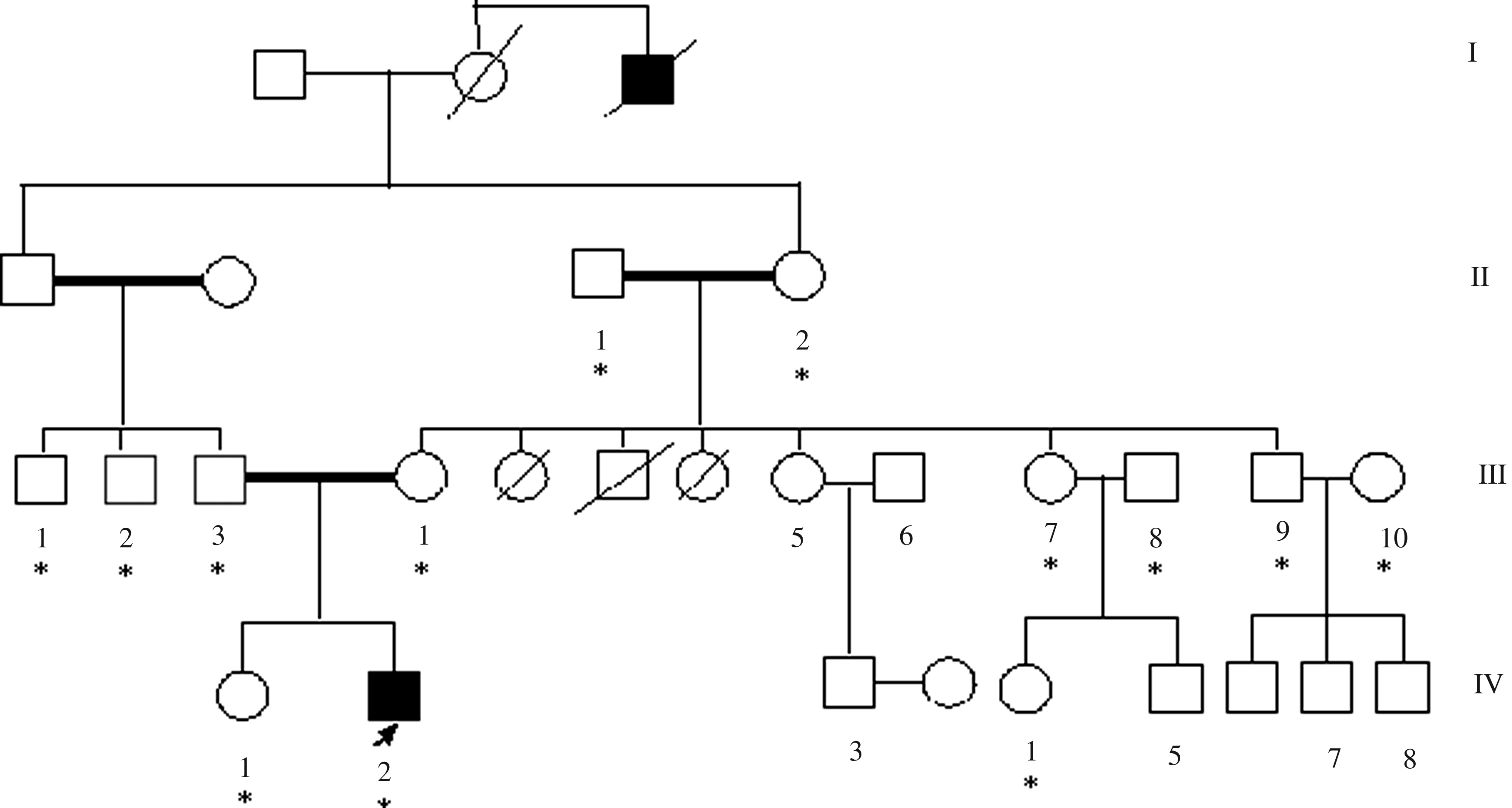
Pedigree of family mt-01 with m.1453A>G (MT-RNR1) and p.W24X (GJB2) mutations. Hearing impaired individuals are indicated by filled symbols. Arrow denotes proband. Asterisks denote the individuals screened for mutations. Genotype (m.1453 A>G/GJB2) of the individuals was as follows: IV-2 (Proband)—G/p.W24X/p.W24X; IV-1 and IV-4—G/Wt/Wt; III-4, III-7, III-9, and II-2—G/p.W24X/Wt; III-1 and III-3—A/p.W24X/Wt; and III-2, III-8, III-10, and II-1—A/Wt/Wt.
Secondary structure effect
It has been previously reported that m.1555A>G alters the secondary structure of the MT-RNR1 molecule (Prezant et al., 1993; Casano et al., 1998). Under this assumption, we analyzed the newly identified sequence changes m.1453A>G, m.1462G>A, and m.1508C>T for their possible effects on secondary structure of the rRNA using the RNAfold software. In this model, the three variants showed deviation from the wild-type prediction and also the m.1555A>G mutation that was much more different in structure (Fig. 2).
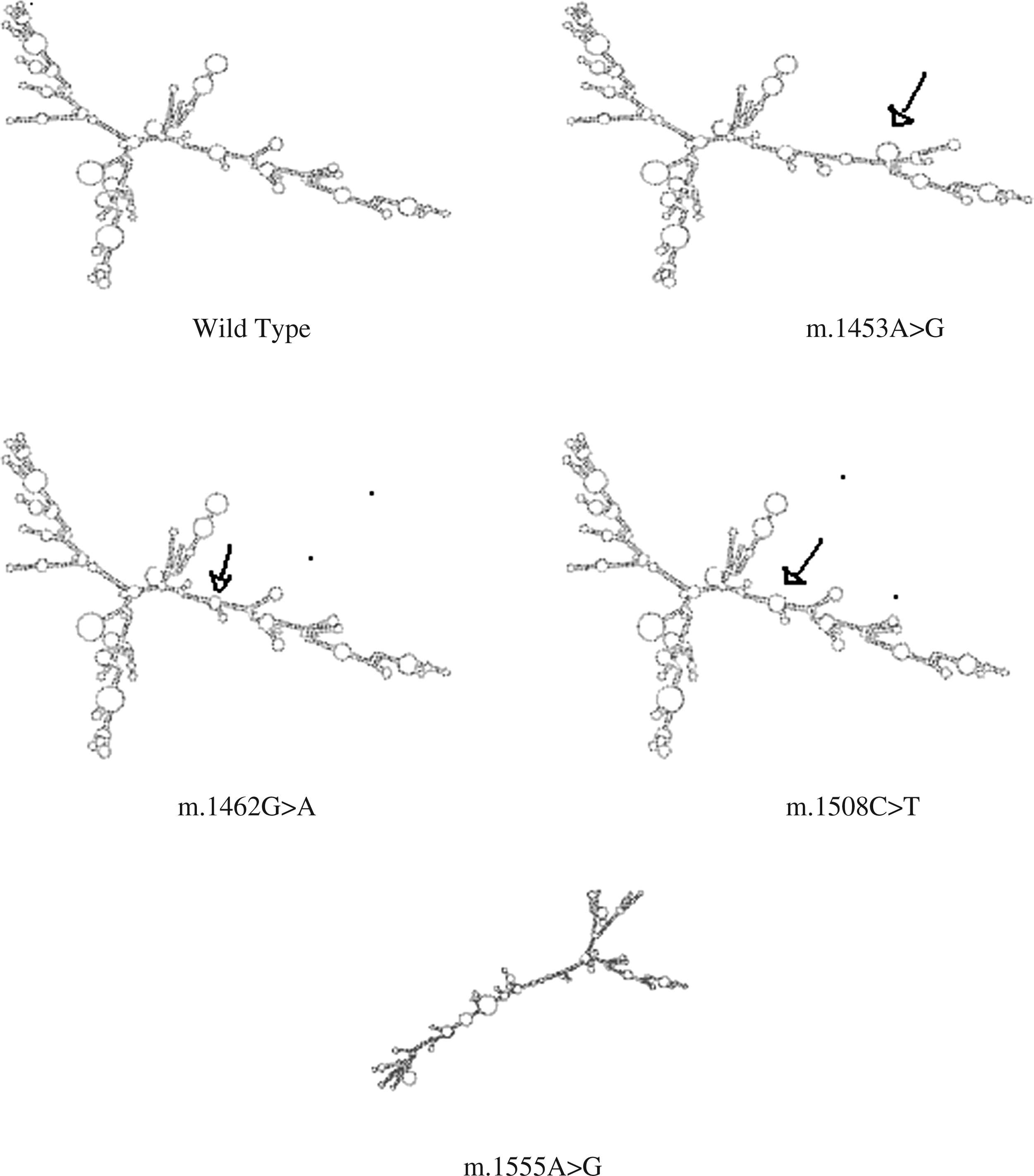
The secondary structures of RNA generated by RNAfold. The deviated regions are marked with arrows. Arrow in m.1453 A>G indicates major difference with reference to wild type. Arrow in m.1462G>A and m.1508C>T indicates minor difference with that of wild type. Whereas m.1555A>G is entirely deviated from that of wild type.
Discussion
Mutations in mtDNA have been found to be one of the most important causes of sensorineural hearing loss (Abe et al., 1998; Achilli et al., 2004). In particular, the homoplasmic m.1555A>G and m.1494 C>T mutations in the highly conserved decoding site of the MT-RNR1 gene have been associated with both aminoglycoside-induced and nonsyndromic hearing impairment in many families world wide. Bhalla et al. (2009) in their study on North Indian patients did not observe any of the known mitochondrial mutations in their cohort. Other studies describing the prevalence of mitochondrial mutations are lacking from the Indian population. Hence, in the present study, we made an attempt to screen for m.1555A>G and other sequence changes in the MT-RNR1 gene in addition to mutations in GJB2 and GJB6 genes in patients with congenital, profound (≥90 dB), nonsyndromic hearing impairment.
In the present study we identified three probands positive for the m.1555A>G mutation, accounting to 1.0% that is comparable with the frequency (0.6-2.5%) reported in other ethnic groups (Usami et al., 2000; Hutchin et al., 2001; Kupka et al., 2002; Tekin et al., 2003; Li et al., 2004; Jacobs et al., 2005).
Since mutations in mtDNA account for a high number of familial and sporadic sensorineural hearing loss cases (Jacobs et al., 2005), we considered the possibility of mutations other than m.1555A>G in MT-RNR1 gene causing nonsyndromic deafness. Our search for other mutations in this gene led to the identification of three novel changes, namely, m.1453A>G, m.1462G>A, and m.1508C>T, in MT-RNR1 gene that were submitted to the Human Mitochondrial Genome Database (http://mitomap.org) with the reference numbers #20081106002, #20081106001, and #20081105001, respectively. The fact that all the three changes were detected only in the patients and not in the controls with altered secondary structure suggests their role in the pathogenesis of the condition.
The m.1453A>G mutation found in the two families (one familial mt-01 and the other sporadic mt-02) was inherited in a typical maternal transmission. However, except for the two probands, none of the family members carrying the mutation showed any traces of hearing impairment. This is expected as a wide variety of symptoms have been reported in deaf patients with mitochondrial mutations, including cases of normal hearing (Fischel-Ghodsian et al., 1993; Hutchin et al., 1993; Prezant et al., 1993; Estivill et al., 1998). Also, previous data suggest that expression of the clinical phenotype of deafness-associated homoplasmic mutations in the MT-RNR1 gene, such as the m.1555A>G (Prezant et al., 1993; Guan et al., 1996; Li et al., 2004; Young et al., 2006; Wang et al., 2008), requires the contribution of modulating factors including environmental factors or nuclear modifier genes.
Previous studies suggested connexin-26 (GJB2) gene as a potential candidate modifier gene wherein several mutations are causally related to hereditary hearing loss (Abe et al., 2001; Friedman and Griffith, 2003). In particular, it has been implied that GJB2 mutations modulate the severity of hearing loss when associated with the mitochondrial MT-RNR1 m.1555A>G mutation (Fischel-Ghodsian, 1999). Hence, we performed mutational screening of the GJB2 gene in probands with m.1555A>G, m.1462A>G, and m.1508C>T and also in members of the two families harboring the m.1453A>G mutation. The analysis revealed segregation of the p.W24X mutation of GJB2 gene in the family (mt-01) harboring the m.1453A>G mutation in the MT-RNR1 gene. In our previous study (Padma et al., 2009) this mutation was found to be the most common allele accounting for nearly 86.7% of the mutated alleles. This mutation was found to be homozygous in 24 and heterozygous in 4 probands with no other detected mutations in Cx26 and hence they were considered as compound heterozygotes with the second allele yet to be explored. Of the seven members of mt-01 carrying the m.1453A>G mutation, the p.W24X mutation in the GJB2 gene was found in a homozygous state in the proband IV-2 (Fig. 1) with profound congenital hearing impairment (100 dB) and in a heterozygous state in III-4, III-7, III-9, and II-2 all of whom had normal hearing. The p.W24X mutation was not detected in IV-1 and IV-4 who carried the mutation m.1453A>G in the MT-RNR1 gene but had normal hearing. It is possible that the occurrence of these two mutations might be an independent phenomenon in this family with the proband being affected with hearing impairment because of the p.W24X mutation alone.
The CI of m.1453A>G was found to be 82%, suggesting it to be a possibly deleterious mutation. Ruiz-Pesini and Wallace (2006) suggested that for a mutation to be deleterious, it should be (1) absent in controls (2), should have a CI>78%, and (3) should have a potential structural and functional alterations. However, the functional role of this mutation has to be elucidated.
In the probands carrying m.1555A>G, m.1462G>A, and m.1508C>T mutations and in sporadic family (mt-02) with the m.1453A>G mutation, mutations in the GJB2 or GJB6 gene were not detected. The defective phenotype in these probands could have arisen due to (1) mitochondrial mutation alone, (2) de novo origin of autosomal dominant mutation irrespective of mitochondrial mutation, (3) other nuclear gene mutation modulating the mitochondrial mutation to cause hearing impairment, or (4) an autosomal recessive mutation irrespective of the mitochondrial variant. The CI of m.1462G>A and m.1508C>T was found to be 64% and 36%, respectively. The m.1462G>A was reported to be a polymorphism by Lu et al. (2010).
mtDNA mutations not only affect tissues with high energy requirements, such as muscle and brain, but also the cochlea. The exact mechanism of cochlear damage in mtDNA-associated disorders is unclear. Normal hearing is dependent upon the hair cells and the stria vascularis, which maintain the ionic gradients necessary for sound signal transduction. Both stria vascularis and hair cells are highly metabolically active and would be compromised by a dysfunction of intracellular mitochondrial ATP as a consequence of mtDNA mutation (Fischel-Ghodsian, 1999).
For the m.1555A>G and m.1494C>T mutations, in which aminoglycoside-induced deafness is believed to be genetically determined, it has been hypothesized that the mutations make the human mitochondrial small rRNA more similar to the bacterial rRNA, the target of aminoglycoside action (Guan, 2004; Qian and Guan, 2009). Accumulation of aminoglycosides in cochlear mitochondria would lead to an inhibition of protein synthesis by interacting with the MT-RNR1 carrying these mutations. As a result of this mitochondrial translation defect, ATP production declines and the generation of reactive oxygen species increases, consequently damaging hair cells and giving rise to hearing impairment. In the absence of aminoglycoside exposure, an analogue mechanism is expected, but the factors leading to a dysfunction of mitochondrial protein synthesis remain unknown. A similar scenario is also possible for the new changes we describe here, in which the phenotypic variability may be explained by the involvement of environmental or genetic factors, contributing to the penetrance of mtDNA mutations. Functional studies using cell lines derived from patients may help in understanding the pathogenic mechanism underlying mitochondrial-mutation-related hearing impairment as studied in the case of the m.1555A>G mutation wherein the phenotypic effects of the mutation were analyzed in lymphoblastoid cell lines derived from the hearing impaired patients (Guan et al., 1996, 2001).
Footnotes
Acknowledgments
The authors thank all the participants for their kind contribution to this study. G. Padma is thankful to Lady Tata Memorial Trust for research fellowship. The authors would like to thank Dr. G. Bhanuprakash Reddy and Ms. Mona Chaurasiya of Bioinformatics Department of National Institute of Nutrition (NIN), for the help rendered by them.
Author Disclosure Statement
No competing financial interests exist.