
Abstract
Aim: Our aim was to examine the diagnostic yield of genetic testing in 855 consecutive unrelated cases referred for Long QT syndrome (LQTS). Results: Eight hundred fifty five consecutive patients with a mean age at testing of 27.5±18.6 years, were referred for LQTS genetic testing and had accompanying clinical information. KCNQ1, KCNH2, SCN5A, ANK2, KCNE1, KCNE2, CACNA1C, KCNJ2, CAV3, and SCN4B were analyzed using Next-Generation sequencing in all patients, and 395 patients were also tested for an additional two genes, AKAP9 and SNTA1. We retrospectively analyzed the diagnostic yield of this genetic test and factors that predicted the likelihood of a disease causing mutation using ANOVA, χ2, t-test, and receiver operator curves. At least one mutation was identified in 30.3% of the patients (n=259), and 18 patients (2.1%) had two mutations. Patients with two mutations had a longer QTc interval (p<0.01) than patients with one mutation. A longer QTc duration and family history of LQTS were each associated with a higher yield of positive results on genetic testing (p<0.01 for each). Using a QTc cutoff of 476 msec or greater, the genetic testing had a sensitivity of 72% and a specificity of 49%. Mutations within the transmembrane domain of KCNQ1 were associated with a greater risk of cardiac arrest and syncope relative to mutations in other domains of the gene. Mutations in SCN5A were associated with a higher frequency of cardiac arrest (52.6%). Conclusion: Sequencing-based genetic testing has a sensitivity of 72% and has clinical utility.
Introduction
C
Materials and Methods
Between 08/05/2008 and 1/1/2011, a total of 855 consecutive unrelated probands with a suspected diagnosis of LQTS were referred for LQTS genetic testing. Clinical data were provided and included age, gender, ethnicity, QTc interval, history of syncope, history of aborted sudden death/sudden death, family history of LQTS, and ICD-9 code. The following LQTS-susceptibility genes were analyzed in all patients: KCNQ1, KCNH2, SCN5A, ANK2, KCNE1, KCNE2, CACNA1C (exons 1-44), KCNJ2, CAV3, and SCN4B. Between 01/14/2010 and 1/1/2011, patients were tested for an additional two genes: (AKAP9, SNTA1) (n=395). All coding regions and splice junctions were PCR amplified and sequenced using the sequencing-by-synthesis method on an Illumina Genome Analyzer for 36 bp reads. The DNA sequence was assembled and analyzed using Mosaik Assembler, v.0.9 (The MarthLab, Boston College), with assembly parameters significantly relaxed for the detection of deletions three to six nucleotides in length. Base calling was done using GigaBayes (The MarthLab, Boston College) in a modified configuration. All potentially disease-associated sequence variants were confirmed and any amplicons with a <20×sequence depth were sequenced by dideoxy sequencing.
A control population of 200 Caucasian and 100 African American subjects was used as a reference population to determine the carrier frequency for any novel variant. Polymorphisms were identified with the Ensembl Genome Browser. Variants were analyzed for predicted functional effect using PolyPhen2, SIFT, Mutation Taster, Softberry Grantham score, cross species alignment, and location compared with published functional domains of the protein. The bioinformatic predictions were used to assess possible pathogenicity based on the type of change (frameshifts, nonsense; missense changes leading to alteration of charge, polarity, and/or size; splicing changes causing exon skipping); evolutionary conservation of the altered position and the region surrounding the change and presence or absence of the variant in large control groups). Variants were classified as mutations if previously reported in the literature as disease causing/associated and if the published data supported that claim, including independent reports, functional data, and segregation data, if the type and location of the variant suggested it had a functional effect based upon informatics predictions, and its absence in controls (Fig. 1). Mutation lists were collated from the published literature, Human Gene Mutation Database, and mutation-specific databases (www.fsm.it/cardmoc/www.lovd.nl/2.0/index_list.php?search_symbol=KCNH2). Variants were classified as likely mutation if there were no published data about the variant, the type and location of the variant suggested it had a functional effect based upon informatics predictions, and it had not been observed in normal controls. Segregation within a family was tested for variants of uncertain significance when possible. Unpublished variants were classified as variants of unknown significance (VUS) when the variant affected a region of the protein not conserved across species, in silico algorithms predicted a benign effect, but the variant is located near published mutations and was not present in ≥1% of control samples. Unpublished variants were classified as a VUS when located in a variable region of the protein, or a region with no known mutations, and bioinformatic prediction models were conflicting or noninformative. Unpublished variants located in alternative transcripts where the gene expression was unknown or when there was insufficient control data from ethnically matched controls were also classified as VUS. Published variants were considered VUS when the reported studies provided insufficient or conflicting clinical and/or functional data. Any published variants with functional studies indicating the variant does not significantly differ from the wild type were classified as VUS. Variants were classified as VUS/likely benign if the type and location of the variant suggested it was unlikely to have a functional effect based upon informatics predictions, and it was observed in <1% of normal controls. Variants were classified as polymorphisms if they were observed in >1% of normal controls.

Algorithm for classification of genetics variants in LQT genes. For novel missense changes, the presence or absence of several major and minor evaluation criteria conjointly determines the final variant classification. *For autosomal dominant genes: missense variants listed in databases with an allele frequency of <1%, with data for at least 200 alleles. ^For novel missense changes, the presence or absence of several major and minor evaluation criteria conjointly determines the final variant classification. VUS, variants of unknown significance.
Based upon the classification above, we placed patients into one of the three groups; (1) no mutation and no VUS, (2) only VUS or VUS/likely benign, and (3) one or more mutations or likely mutation variants. Patients with only VUS were categorized in the VUS group, including patients with multiple VUS. Patients with both a mutation and a VUS were placed in the mutation group.
The transmembrane region of the KCNQ1-encoded channel was defined as the coding sequence involving amino acid residues from 120 through 355, with the N-terminus region defined before residue 120 and the C-terminus region after residue 355. The transmembrane region of the KCNH2-encoded protein was defined as the coding sequence involving amino acid residues from 404 through 659 (the pore loop region: 548-659), with the N-terminus region defined before residue 404, and the C-terminus region after residue 659.
Categorical variables are presented as counts and percentages, and were compared using Chi-square analysis. Continuous variables are presented as means and standard deviations and were compared using the t-test or ANOVA, as appropriate. Receiver operator curves (ROC) were used to determine the optimal cutoff of QTc duration for genetic testing. p<0.05 was considered significant. ROC analysis was performed using PASW Statistics 18 (Chicago, IL).
Results
Patient characteristics
Table 1 summarizes the characteristics of this cohort. Five hundred fifty five females (64.9%) and 298 males (34.9%) were referred for testing. Sex was not reported for two patients (0.2%). Ethnicity was reported for 594 (69.5%) of the patients: 460 (77.4%) Caucasian, 29 (4.9%) African American, 31 (5.2%) Hispanic, 30 (5.0%) Asian, 6 (1.0%) Pacific Islander, 8 (1.3%) Native American, 5 (0.8%) Middle Eastern, and 25 (4.2%) mixed ethnicity. The mean age at testing was 27.5±18.6 years (range 0 to 92 years of age); 49.5% of the patients were younger than 18 years of age. Male probands were younger at testing compared with female probands (23.6±18.7 vs. 29.7±18.2 years, p<0.001). One hundred and eighty three (61.6%) of the males were younger than 18 at the time of testing compared to 237 (42.9%) of the females (p<0.001).
p<0.05 three-way comparison; **p<0.01 three-way comparison; #p<0.01 mutation versus VUS; ##p<0.05 mutation versus VUS; +p<0.01 mutation versus no mutation; ++p<0.05 mutation versus no mutation; ^Total cohort number is 855. Since data were not reported for all patients, for all data, the number of patients with reported data for the variable is indicated in the denominator.
VUS, variants of unknown significance.
Clinical correlates with genetic test results
Overall, 259 of 855 (30.3%) patients had a positive genetic test result with either a mutation or likely mutation identified (Table 1). The yield of testing was similar in males and females (p=0.52). The age at testing was not associated with a different yield (p=0.23). A hearing loss was reported in 12 patients with a mean QTc of 477 msec, but none of them had two mutations in KCNQ1 or KCNE2 associated with the Jervell and Lange-Nielsen syndrome. Non-Caucasians were no more likely to have VUS than Caucasians (p=0.31).
QTc was reported for 569 patients (66.6%). Average QTc was 490±54 msec, 57.6% of the QTc intervals were higher than 475 msec. Mean QTc for patients with a mutation was 509±61 msec; 476±46 msec for patients with a VUS; and 483±49 msec for patients with no mutation (Table 1 and Fig. 2). A longer QTc duration was associated with a higher yield of genetic testing (p<0.01). Using a QTc cutoff of 476 msec, genetic testing had a sensitivity of 72% and a specificity of 49%. Data on syncope were reported for 614 (72.0%) patients, and 308 (50.2%) of these patients had a reported syncopal episode. A history of syncope was not associated with a higher yield of genetic testing. A family history of LQTS was reported for 592 (69.2%) cases and was positive for 294 (49.7%) of these cases and was associated with a higher yield of genetic testing (p<0.01). Information on sudden cardiac death/aborted cardiac arrest in the patient was documented for 627 cases (73.3%), was positive for 190 (30.3%) of these patients, and was associated with a lower yield of genetic testing (p=0.03).
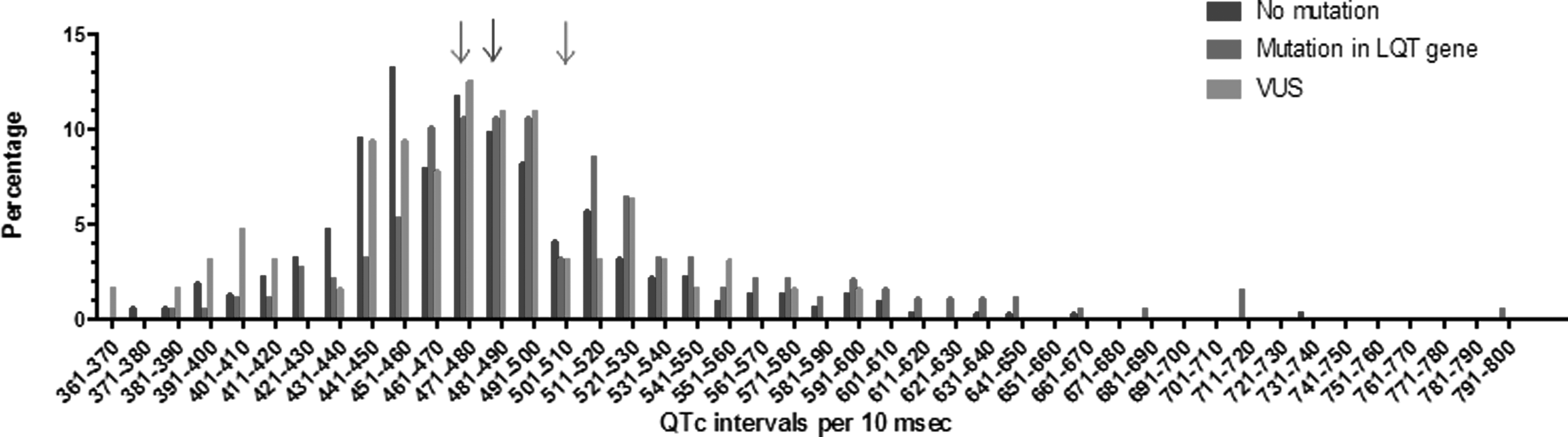
Distribution of QTc by LQT genetic test result. Mean QTc for each genetic test result indicated with an arrow: 483 msec for no mutation, 477 msec for VUS, and 508 msec for mutation-positive.
Clinical characteristics by number of mutations
Among the 259 mutation-positive patients, 93.1% had a single mutation with the following distribution of mutations: 114 (47.3%) KCNQ1, 81 (33.6%) KCNH2, 28 (11.6%) SCN5A, 1 (0.4%) ANK2, 6 (2.5%) KCNE1, 3 (1.2%) KCNE2, 6 (2.5%) KCNJ2, and 2 (0.8%) CACNA1C (Fig. 3). The other 18 patients (2.1% of total cohort and 6.9% of patients with a positive result) had two mutations (Table 2). Multiple mutations in the same gene were observed in five patients. There was no difference in gender distribution or age at diagnosis between patients with one mutation and patients with two mutations. Patients with two mutations were no more likely than those with one mutation to have syncope (p=0.75), sudden cardiac death/aborted cardiac arrest (p=0.38), or a positive family history (p=0.90). Patients with two mutations had a longer QTc (505±57 msec for one mutation versus 551±98 msec for two mutations, p<0.01).
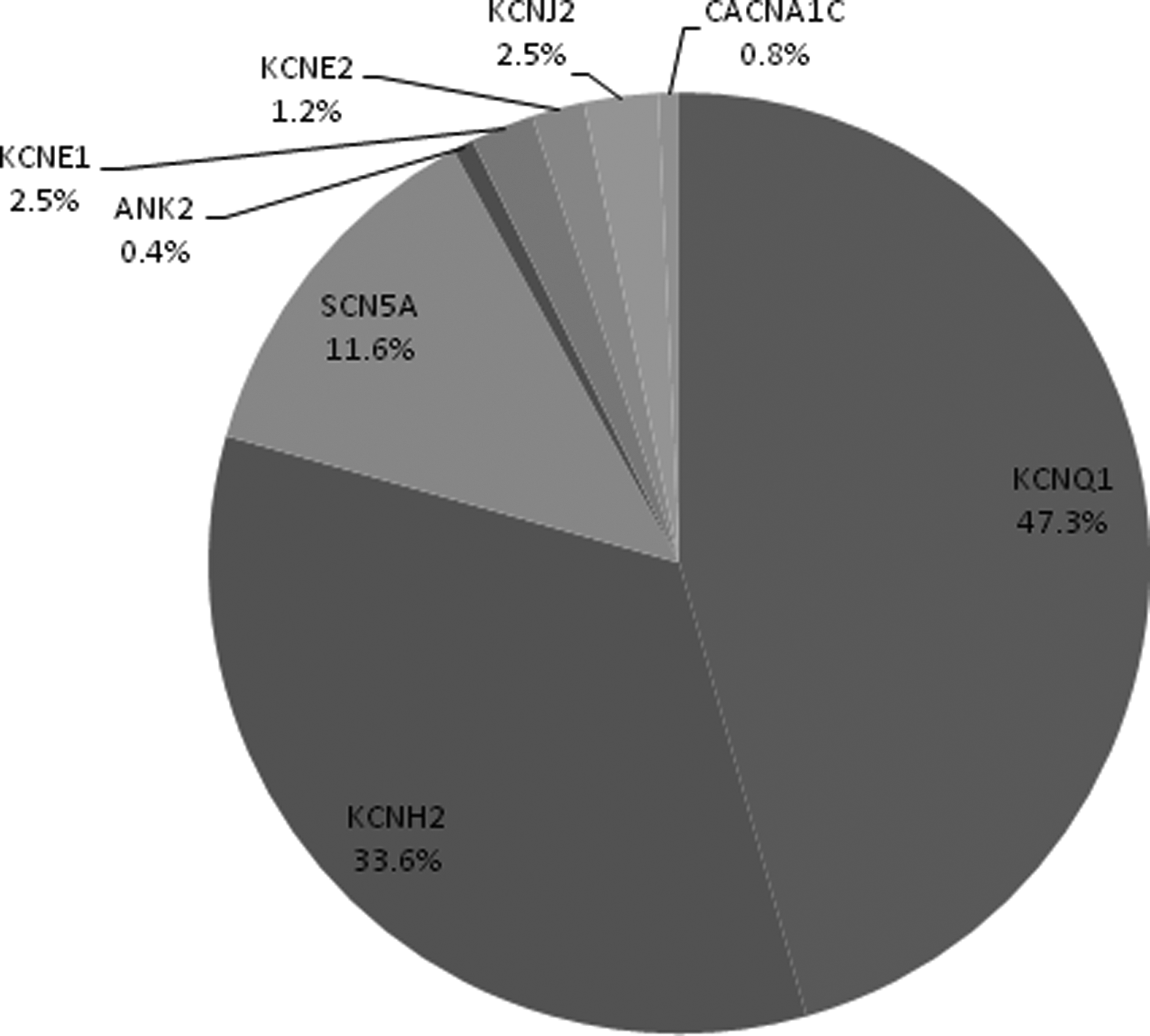
Distribution of LQT mutations by gene.
Mutations identified
We identified 277 mutations and 87 VUS (Figs. 3 and 4). Seventy eight (40.4%) of the mutations identified were novel (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/gtmb). The distribution of mutation types was 216 (78.0%) missense, 23 (8.3%) nonsense, 18 (6.5%) splice site, and 20 (7.2%) frameshift (Supplementary Table S1). There was no difference in the clinical characteristics of individuals who had missense mutations versus non-missense mutations for any of the genes.
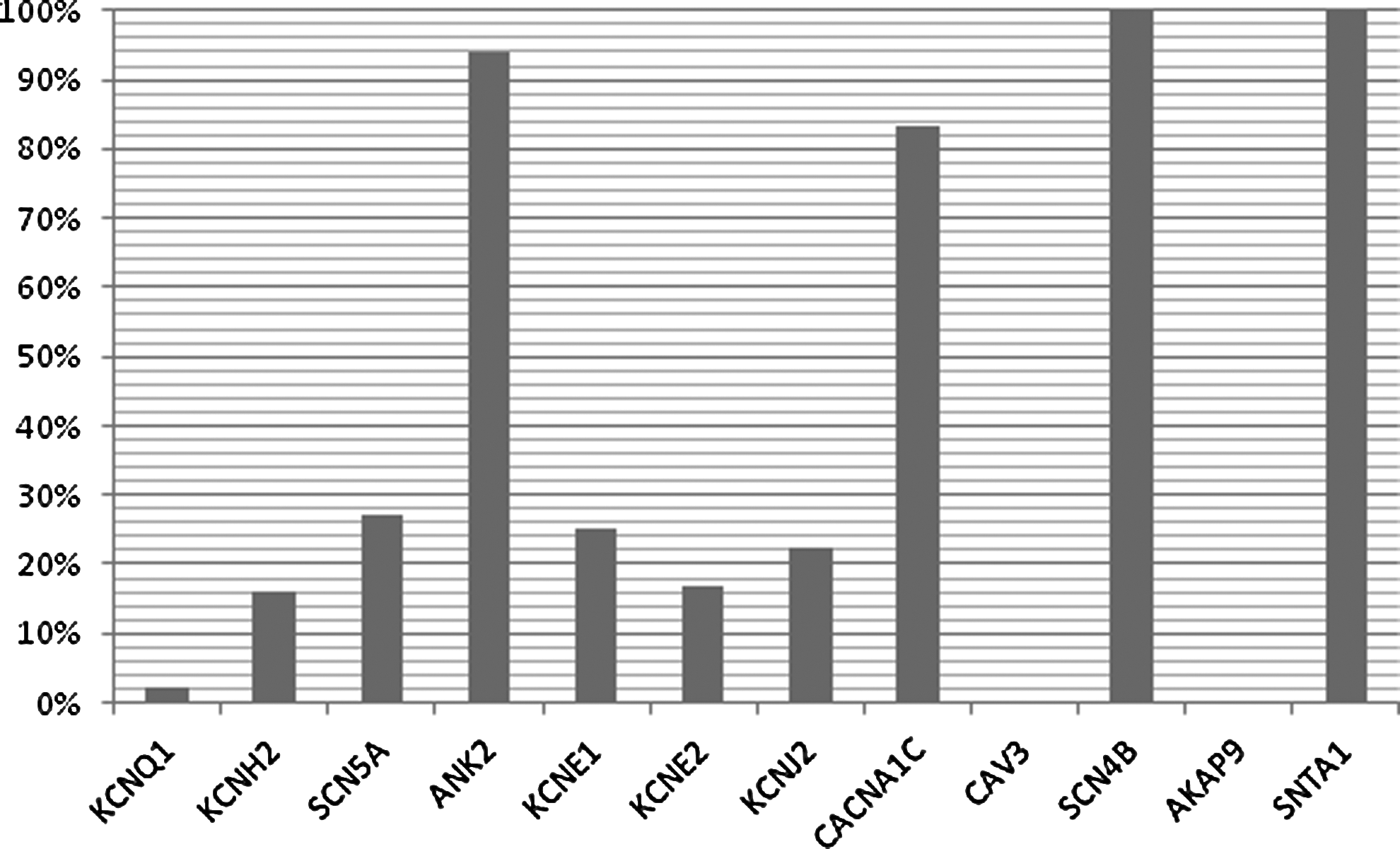
Percentage of VUS identified as percentage of all rare variants identified by gene.
Clinical characteristics by gene and mutation location
In patients with only one LQTS mutation, mean QTc durations were similar if the mutation was in KCNQ1, KCNH2, or SCN5A (513±52 msec vs. 497±53 msec vs. 509±68 msec, p=0.27) (Table 3). Patients with a SCN5A mutation were more likely to have an episode of cardiac arrest (13.4% in KCNQ1, 33.3% in KCNH2, and 52.6% in SCN5A) compared with patients with a mutation in KCNQ1 (p=0.02) or in the three-way comparison (p<0.01). Also, patients with a mutation in KCNH2 were more likely to have an episode of cardiac arrest compared to patients with a mutation in KCNQ1 (p<0.01) (Table 3). However, we did not find any significant difference in the family history, syncope, or age at testing between mutation carriers for KCNQ1, KCNH2, and SCN5A (Table 3).
p<0.01 for comparison between KCNQ1 and SCN5A; #p<0.01 for three-way comparison between KCNQ1, KCNH2, and SCN5A; ^p<0.01 for comparison between KCNQ1 and KCNH2.
SCD, sudden cardiac death.
When comparing the locations of the KCNQ1 mutations, we found that individuals with KCNQ1 mutations in the transmembrane domain had significantly more cardiac arrests (p=0.02) and syncopal episodes (p<0.01) than those whose KCNQ1 mutations were in the N/C terminus (Table 4A). However, we found no significant difference in the QTc for patients with a KCNQ1 transmembrane domain (516±63 msec) compared to a N/C terminus mutation (507±35 msec) (p=0.46).
When analyzing patients with only one KCNH2 mutation, we did not find any difference in the frequency of syncope (p=0.08) or cardiac arrest (p=0.16) between missense versus non-missense mutations. When correlating a mutation with clinical information, we did not find a difference in syncope, cardiac arrest, or QTc for transmembrane versus transmembrane-pore domain, N-terminus or C-terminus to each other (Table 4B).
NS, not significant.
A total of 87 VUS were identified. The percentage VUS of all the variants identified by gene was 3 (2.3%) in KCNQ1, 18 (16.2%) in KCNH2, 13 (27.1%) in SCN5A, 30 (93.8%) in ANK2, 2 (25.0%) in KCNE1, 1 (16.7%) in KCNE2, 2 (22.2%) in KCNJ2, 10 (83.3%) in CACNA1C, 5 (100%) in SCN4B, and 3 (100%) in SNTA1 (Fig. 4). One or more VUS were identified in 82 patients (9.6%). Of VUS, 61 (74.4%) were classified as of uncertain significance and 21 (25.6%) as likely benign. For the VUS in ANK2, CACNA1C, AKAP9, and SNTA1, we found that patients harboring one of these VUS were clinically not significantly different from patients with no mutation/VUS (Table 5).
Discussion
Genetic testing for LQTS is now widely available and is increasingly utilized clinically. Our series of 855 unrelated patients demonstrated a yield of 30.3% when testing for a panel of 12 genes for LQTS in a population with a mean QTc of 490±54 msec. This study presents data on the yield of genetic testing for the largest number of LQT genes currently available in a clinical diagnostic setting. A few studies have reported on the yield of Long QT testing previously; however, these studies were limited to five genes (KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2) (Splawski et al., 2000; Tester et al., 2005; Kapplinger et al., 2009).
Our series demonstrated a yield of 30.3%, suggesting that physicians request testing for some patients with an intermediate pretest probability. Reasons for test submission by the referring physician are not always known, but in many cases, physicians may be seeking additional data to further risk stratify patients with borderline QTc intervals without other clinical features to evaluate a LQT diagnosis. As expected, a longer QTc duration was associated with a higher yield of genetic testing. Using a QTc cutoff of 476 msec, the genetic testing panel had a sensitivity of 72% and a specificity of 49%. These results are comparable to the results from the series from another clinical laboratory performing genetic testing for five genes with a positive yield of 36%; however, no patient characteristics were available for that cohort to determine if the clinical cohorts are comparable (Kapplinger et al., 2009). In testing of research cohorts, the clinical definition of LQTS was more stringent, and yield of genetic testing ranged from 44%-68% (Splawski et al., 2000; Napolitano et al., 2005; Tester et al., 2005). Tester et al. showed that the yield of genetic testing for the five most common genes was nearly 75% among patients with the highest clinical likelihood for LQTS (Schwartz-Moss score ≥4), while the yield in the entire research cohort with a mean QTc 482±57 msec was 50% (Tester et al., 2005). The distribution of the mutations across genes found in our study was comparable to other studies with the majority of mutations in KCNQ1 (47.3%), KCNH2 (33.6%), and SCN5A (11.6%) (Splawski et al., 2000; Napolitano et al., 2005; Kapplinger et al., 2009). Other series have reported that of mutation-positive patients, the range of the distribution of mutations by a gene is 42%-49% for KCNQ1, 32%-45% for KCNH2, and 8%-13% for SCN5A (Splawski et al., 2000; Napolitano et al., 2005; Tester et al., 2005; Kapplinger et al., 2009). Patients in our series with multiple mutations were found to be younger at testing and have a longer QTc, consistent with previous studies (Westenskow et al., 2004; Tester et al., 2005).
About 55.1% of all the VUS identified were found in ANK2, CACNA1C, SCN4B, and SNTA1, whereas only 1.4% of mutations were identified in these four genes. This is, in part, because there are less published data about mutations in these genes to guide interpretation of novel rare variants. We found that the clinical characteristics of the patients with VUS in ANK2, CACNA1C, AKAP9, and SNTA1 were not significantly different from patients with no VUS/mutations. More functional data about these genetic variants would greatly enhance future decisions about the clinical utility of including these four genes in LQT gene testing panels. An optimized LQT panel would decrease the confusion and ambiguity that may be associated with VUS for providers and patients. Comparatively, the percentage of mutation-positive patients was 28.2% for LQTS1-3, 29.3% for LQTS1-6, and 30.3% for LQTS1-12. Increasing the number of genes tested increases the sensitivity of the test, but this should be considered within the context of the pretest probability of carrying a mutation given the clinical scenario and balanced with the potential confusion of returning a VUS.
The distribution of mutation types in our series was 78.0% missense, 8.3% nonsense, 6.5% splice site, and 7.2% frameshift. When comparing the mutation types to other series, we find similar distributions by mutation types: 70%-72% missense, 5%-6% nonsense, 3%-7% splice site, and 10%-15% frameshift (Splawski et al., 2000; Napolitano et al., 2005; Kapplinger et al., 2009). The mutation type did not correlate with clinical outcome for any of the LQT genes. However, when comparing the locations of mutations in the KCNQ1 gene, we found that patients with KCNQ1 transmembrane mutations and specifically pore mutations had significantly more cardiac arrests and syncopal episodes than KCNQ1 mutations in the C-terminus, similar to results by Moss et al. (2007). Recently, Basheshet et al. showed us that patients with a KCNQ1 C-loop missense mutation were at increased risk for life-threatening events (Barsheshet et al., 2012). However, our data did not show such an increased risk for life-threatening events possibly due to limited statistical power. For KCNH2, we were not able to demonstrate a relation between the location of the mutation and clinical parameters, probably due to our limited sample size of 47 mutation-positive patients with complete clinical information.
Our series potentially represents a less biased cohort compared with previous research studies, since our series includes only probands and is not enriched for families highly motivated to enter research studies based upon a strong family history. Patients and families identified through clinical testing will provide an important opportunity to study the full range of the LQTS phenotype and hopefully provide better estimates of penetrance and risk of cardiac arrest to guide management decisions.
Study limitations
The major study limitation is the incomplete provision of clinical information on the testing requisition forms. QTc durations were provided for 66.6% of the patients and sudden cardiac death/aborted cardiac arrest for 84.4%. ECGs and medical records were not routinely available for confirmation. The family history was available for 69.2%, syncope for 69.9%, and ethnicity for 69.5%. Data on age at testing, genetic diagnosis, and gender were reported in nearly all patients. Consequently, clinical correlations with the genetic test results were only possible for 72% of patients, limiting the power of our analyses.
Conclusion
This study provides a summary of genetic testing results from a clinical population evaluated for LQTS. Factors that were most predictive in identifying a LQTS mutation were the family history and QTc duration. The sensitivity of the test was 72% for patients with a QTc>476 msec. Genetic testing identifies the gene and mutation location, which can have clinical implications for the patient and provides a reliable method to test family members and therefore has a clinical utility.
Footnotes
Acknowledgments
The authors acknowledge the valuable contribution of Sonia Benhamed and the other laboratory staff at GeneDx for their valuable contribution. The authors also acknowledge the contribution of all the referring physicians.
Author Disclosure Statement
Leah Williams, Amy Daly, Gabriele Richard, Sherri Bale, and Daniela Macaya are employees of GeneDx.
Wendy K. Chung is a consultant for GeneDx.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.