
Abstract
Background: Hereditary hemorrhagic telangiectasia (HHT) is an autosomal-dominant vascular disorder with three disease-causing genes identified to date: ENG, ACVRL1, and SMAD4. We report an HHT patient with allelic dropout that on routine sequence analysis for a known mutation in the family (c.817-3T>G in ENG) initially seemed to be homozygous for the mutation. Aim: To explore the possibility of allelic dropout causing a false result in this patient. Methods: Mutation analysis of additional family members was performed and haplotype analysis carried out. New primers were designed to reveal the presence of a possible sequence variant, which could explain the presumed allelic dropout. Results: Allelic dropout caused by a six-nucleotide duplication close to the standard reverse primer was the assumed cause of a false homozygous diagnosis. Conclusion: Sequence variants outside of the primer regions can be the cause of allelic dropout, creating unforeseen errors in genotyping. Our finding emphasizes the need for careful quality control in all molecular genetic studies.
Introduction
The clinical diagnosis of HHT is established based on the Curaçao criteria. Approximately 85% of HHT patients (Brusgaard et al., 2004; Lesca et al., 2004; Schulte et al., 2005) have a mutation in either ENG, ACVRL1, or SMAD4.
During the genotype analysis of an HHT family, an affected female family member appeared homozygous for the familial splice-site mutation in ENG. Homozygosity for HHT-causing mutations is believed to be lethal in utero (Bourdeau et al., 1999; Li et al., 1999; Arthur et al., 2000), leaving two scenarios to explain this finding. First, our patient could in fact be homozygous and the mutation a benign variant. Second, the result of the genetic analysis could be artificial, obscuring the presence of a normal allele.
The 17-year-old daughter of our patient did not carry the mutation, as would be expected if in fact the mother was homozygous for the mutation. In addition, the daughter did not show any clinical signs of HHT: no epistaxis, telangiectasia, nor pulmonary arteriovenous malformation (PAVM).
We hypothesized that failure to detect the wild-type allele by sequence analysis, causing a false homozygous result in the female, could be due to allelic dropout (nonamplification) of the paternal allele. To investigate the reason for this, we performed haplotype analysis using seven microsatellites, and we analyzed for sequence variants in and around the primer-binding sites.
Materials and Methods
Patients
The pedigree of the family is shown in Figure 1 and described in Table 1. The HHT diagnoses in the family members are based on the Curaçao criteria (Shovlin et al., 2000). The married couple in generation II is unrelated; she is Danish (Caucasian), and he originates from Indonesia (Austronesian).
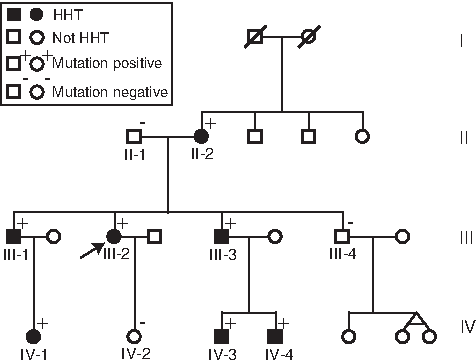
Pedigree of the family. Arrow marks our patient. HHT, hereditary hemorrhagic telangiectasia.
Contrast echocardiography indicated a right-to-left shunt in the lungs, and multislice CT raised suspicion of a very small PAVM.
Contrast echocardiography indicated a right-to-left shunt in the lungs, but multislice CT identified no PAVM.
+, fulfilling the Curaçao criteria; −, not fulfilling the Curaçao criteria; HHT, hereditary hemorrhagic telangiectasia; PAVM, pulmonary arteriovenous malformation.
The female patient (III-2, Fig. 1) was analyzed at the age of 48 and had epistaxis three to four times a week and telangiectasia present in the nasal and oral cavities. Contrast echocardiography (Andersen and Kjeldsen, 2010) indicated a right-to-left shunt in the lungs. Multislice CAT (CT) scan showed a possible, but very small, PAVM.
The familial mutation was located in the proband of the family (IV-3), her 19-year-old nephew, who presented with PAVM, frequent epistaxis, and telangiectatic lesions in the nasal and oral cavities.
Methods
Genotyping
Standard screening of a proband: Genomic DNA was isolated from peripheral leukocytes using a Maxwell®16 (Promega) robot. All exons and exon-intron boundaries of ENG (accession no. NM_001114753.1) and ACVRL1 (accession no. NM_000020.2) were analyzed by bidirectional sequencing using the BigDye® Terminator v.31 cycle sequencing kit (Applied Biosystems) and an ABI3730XL capillary sequencer (Applied Biosystems). The result was confirmed on an independently collected blood sample. The two genes were also analyzed by Multiplex Ligation-dependent Probe Amplification (MLPA) (SALSA MLPA Kit P093 HHT/PPHI; MRC Holland).
Genomic DNA was isolated from peripheral leukocytes using standard procedures. Exon 7 of ENG and the corresponding exon-intron boundaries were analyzed by bidirectional sequencing for the presence of the familial mutation with the forward primer E7F: 5′-CTGGCATAACCCTGGCTG-3′ (Chr9: 130587331-130587314, according to HG19) and the reverse primer E7R: 5′-ACACAGAGGTGCTTCACCAAC-3′ (Chr9: 130587018-130587998, according to HG19). The results were confirmed on an independently collected blood sample.
Repeated genotyping with new primers was performed on genomic DNA from our patient, her daughter, and both her parents. The new primers for exon 7 of ENG, E7′F: 5′-GGCTCATCGACGCCAACCACAACA-3′ (Chr: 130587521-130587544, according to HG19) and E7′R: 5′-GGCGTCGTCGGCACACTTTGT-3′ (Chr: 130586616-130586636, according to HG19), were designed external to the original primers, to reveal the presence of a single-nucleotide polymorphism (SNP) or intronic sequence variants that could explain the presumed allelic dropout.
Haplotype analysis
Haplotype analysis was performed to determine the phase. Seven microsatellites on chromosome 9 surrounding the Endoglin gene were used. Four located upstream (D9S1850, D9S1798, D9S282, and D9S1821), and three downstream from the gene (D9S1827, D9S159, and D9S1831) (Fig. 2). Haplotype analysis was performed using standard procedures. Microsatellites were separated on an ABI3730XL and analyzed using the GeneMapper® Software v.4.1 (Applied Biosystems).
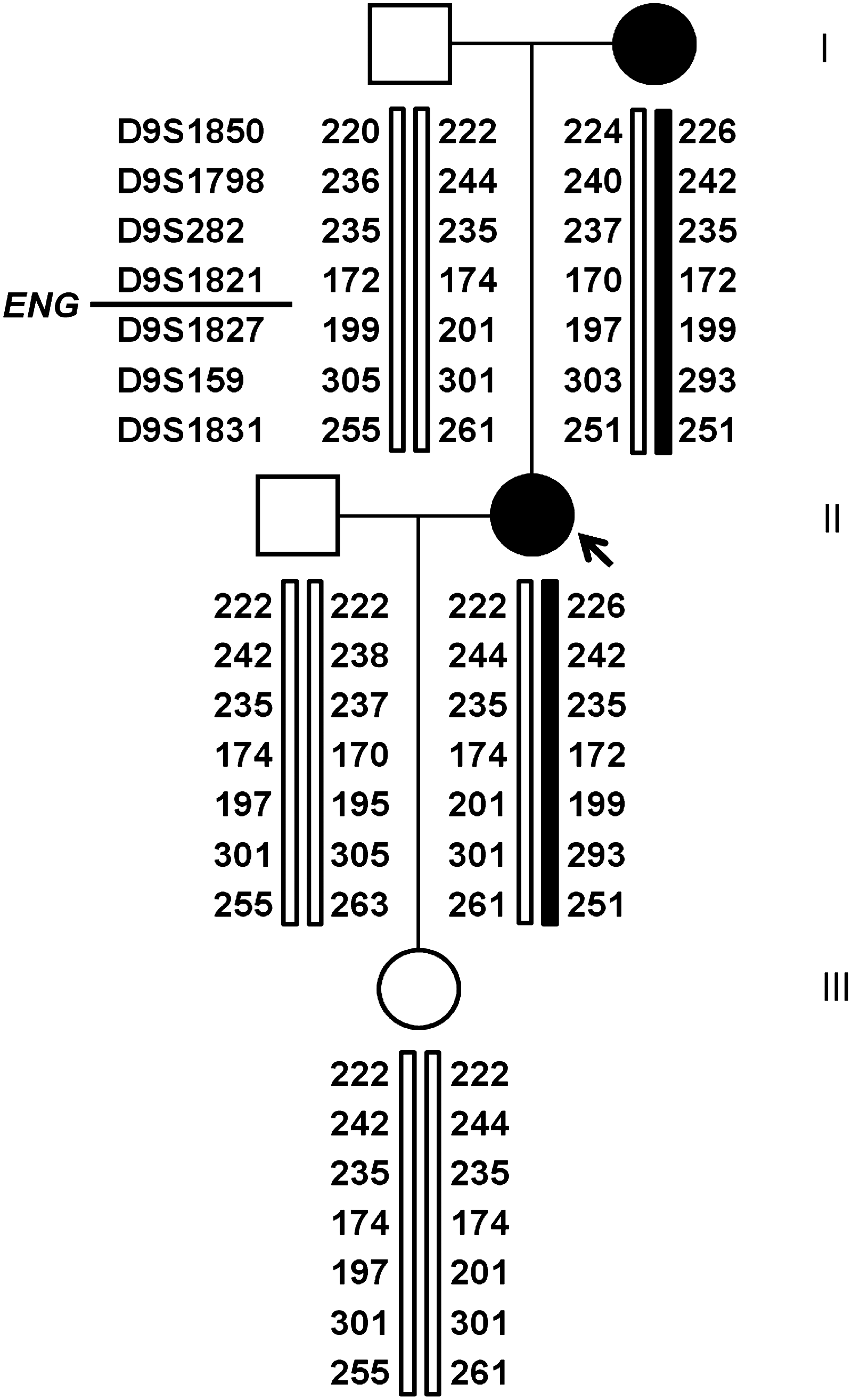
Our patient (arrow), shown here in generation II, has inherited one allele from each parent, with no sign of recombination or crossing-over. The wild-type (white) allele is inherited from the father, and the mutated (black) allele from the mother.
Results
Genotyping
c.817-3T>G in ENG was detected in this family. No other mutation or exonic copy number variation was identified in the family. c.817-3T>G was not previously described in dbSNP build 132, HHT Mutation Database, or HGMD, nor detected in more than 175 patients screened for mutations in our laboratory. However, the mutation segregated with the HHT phenotype in the family (Table 1) and was predicted to be disease causing, based on mutation prediction analysis, using splice-site prediction software. Splice-site Prediction by Neural Networks (www.fruitfly.org/seq_tools/splice.html) predicted c.817-3T to be a weak acceptor with a score of 0.69 and not recognized when substituted by c.817-3G. Using GeneSplicer (www.cbib.umd.edu/software/genesplicer/gene-spl.shtml), the c.817-3T site was only recognized with a sensitivity set point below 60%, and again the c.817-3G was not recognized as an acceptor site.
Haplotype analysis
The haplotype analysis showed that our patient had inherited two different alleles (Fig. 2). The mutated allele was inherited from her mother, and the wild-type allele was inherited from her father, and passed on to her daughter (Fig. 2). No recombination was detected in the region.
Repeated genotyping with new primers
Repeated sequencing with new primers was performed on samples from our patient, her parents, and her daughter. Our patient appeared, as expected, to be heterozygous when genotyping with the new primers (Fig. 3).
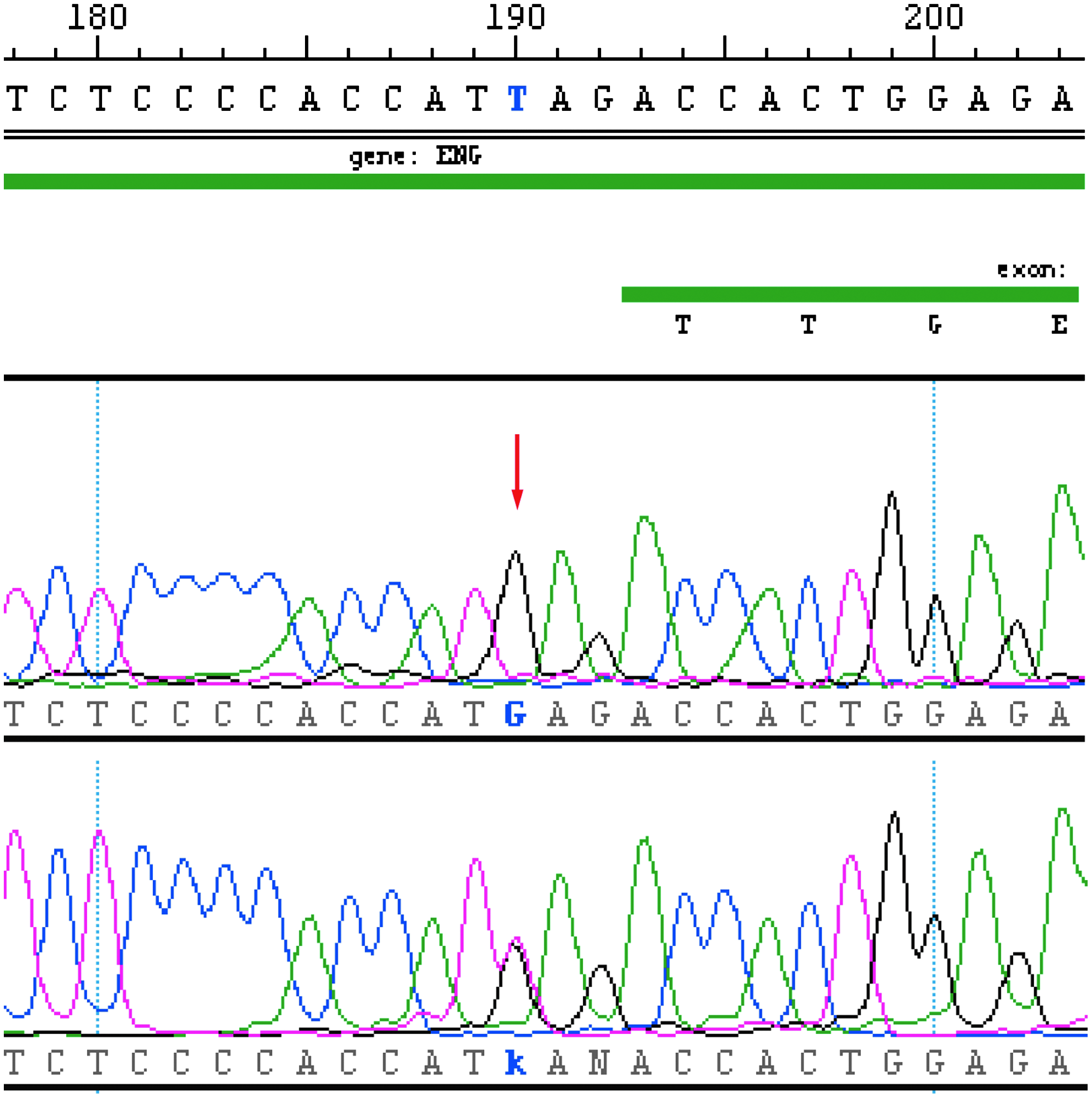
Sequencing results when analyzing for the mutation c.817-3T>G in ENG. With standard primers, a homozygous state (G) is seen in our patient (arrow). With the new primers, a heterozygous state (T>G) in the same position is observed (below). The wild-type sequence is given at the top. Color images available online at www.liebertpub.com/gtmb
No SNP was detected in the original primer-binding sites of our patient, but a six-nucleotide duplication (c.991+21_26dupCCTCCC) situated 34 nucleotides upstream of the 5′-end of the original reverse primer-binding site (Fig. 4) was identified. The duplication was observed in our patient, her father, and her daughter. Similar variants in the same intron have been reported by Lesca et al. (2004), c.992+26insCCTCC, and Lenato et al. (2006), c.991+25_991+26insCCTCC. The UCSC genome Browser, using the dbSNP135 dataset, did not reveal similar or identical variants.

Partial sequence of the ENG gene with exons 6, 7, and 8. Exon and intron sequences are shown in upper- and lowercase, respectively. The splice-site mutation (c.817-3T>G) and the duplication (c.991+21_26dupCCTCCC) are shown in bold. The primer-binding sites of the original primers are underlined (intron sequence). The primer-binding sites of the new primers are shown in italics and underlined (exon sequence).
This duplication was the only sequence variant observed, when sequencing with the new primers, and we assumed this to be the cause of allelic dropout when using our original primers.
To evaluate the prevalence of this duplication in the Caucasian population, we screened 171 Danish healthy controls, with the new primer set. Thirty-seven (21.6%) carried the duplication. When sequencing those 37 with our original primers, none of them showed the duplication, which strongly suggests that the described duplication leads to allelic dropout of the allele in cis.
Discussion
We present here a case of allelic dropout in an HHT patient that could have lead to a false diagnosis. It is most likely caused by the presence of a duplication (c.991+21_26dupCCTCCC) in ENG situated 34 nucleotides upstream of the 5′-end of our original reverse primer-binding site. This was supported by the result of the sequencing of 37 healthy controls carrying the duplication (c.991+21_26dupCCTCCC) in ENG with the new primer set. None of them showed the duplication, using our original primers, which strongly suggests that the described duplication is in fact the explanation of allelic dropout in this case.
SNP in the primer-binding sites as a cause of allelic dropout has previously been reported in relation to a number of other genes (Ellard et al., 1999; Tester et al., 2006; Laios and Glynou, 2008), but was not found in this case. Day et al. (1996) argue for possible allelic dropout in PCR-based analyses of the CYP21 genes in congenital adrenal hyperplasia, but do not localize any sequence variants or other explanations. Allelic dropout, caused by sequence variants outside the primer-binding sites, is, to our knowledge, not previously reported.
The exact reason why a sequence variant outside the primer-binding sites leads to nonamplification is obscure. We can only hypothesize that it could be caused by a different, and more complex, folding of the nonamplified fragment. It should be mentioned though that applying mFOLD (http://mfold.rna.albany.edu/?q=mfold/DNA-Folding-Form) did not point to any changed folding patterns due to the duplication.
This exact duplication is not previously reported (however, similar variants in the same intron have been reported as mentioned above), which might indicate that other laboratories may experience allelic dropout in this region of ENG as well. Other HHT researchers (McAllister et al., 1994; Gallione et al., 1998; Lesca et al., 2004; Lenato et al., 2006) have published their primers for the amplification of ENG exon 7. They are not exactly identical to our original primers, but lie very close, and are situated in the region between our original reverse primer-binding site and the duplication. The prevalence of the duplication in ENG (c.991+21_26dupCCTCCC) is 21.6% in the Danish population, which is surprisingly common. In this case, our patient inherited the duplication from an Indonesian father, suggesting that it is not only a common variant in Caucasians.
The frequency of this variant in ENG, which we believe is the cause of allelic dropout, is alarming, as it may lead to incorrect genotyping in HHT—patients carrying the duplication—and probably not only in our laboratory.
Genotyping of relatives to HHT patients is commonly used presymptomatically, to ensure early screening for internal AVMs in affected individuals. In that case, misgenotyping of an HHT patient as homozygous wild type is potentially lethal.
One way to avoid misgenotyping owing to allelic dropout would be to use two primer sets. This will, however, not be the standard solution in clinical laboratories. The strategy would be obvious in cases where the genotype and phenotype do not match. It may also be considered in cases of known familial mutations in which a correct genotype is crucial, for example, in prenatal diagnostics or testing before important surveillance.
Conclusion
A sequence variant outside the primer-binding sites has in this case been demonstrated to be the likely cause of allelic dropout, creating unforeseen errors in genotyping. When further analyses were performed, this variant turned out to be very common in, at least, the Danish population. This result emphasizes the need for careful quality control in all molecular genetic studies.
Allelic dropout is an important issue and a potential problem underlying a number of false-negative and false-positive results in genetic testing of many disorders.
To our knowledge, this is the first report of allelic dropout due to a common variant, leading to incorrect genotyping in the ENG gene.
Footnotes
Acknowledgments
The authors wish to thank Jette Moeller and Pernille Jordan for technical assistance and Lotte Nylandsted Krogh for helpful discussions.
Author Disclosure Statement
No competing financial interests exist. The authors declare that they do not have any conflict of interest.