
Abstract
Allele-specific amplification on the basis of polymerase chain reaction (PCR) has been widely used for single-nucleotide polymorphism (SNP) genotyping. However, the extraction of PCR-compatible genomic DNA from whole blood is usually required. This process is complicated and tedious, and is prone to cause cross-contamination between samples. To facilitate direct PCR amplification from whole blood without the extraction of genomic DNA, we optimized the pH value of PCR solution and the concentrations of magnesium ions and facilitator glycerol. Then, we developed multiplex allele-specific amplifications from whole blood and applied them to a case-control study. In this study, we successfully established triplex, five-plex, and eight-plex allele-specific amplifications from whole blood for determining the distribution of genotypes and alleles of 14 polymorphisms in 97 gastric cancer patients and 141 healthy controls. Statistical analysis results showed significant association of SNPs rs9344, rs1799931, and rs1800629 with the risk of gastric cancer. This method is accurate, time-saving, cost-effective, and easy-to-do, especially suitable for clinical prediction of disease susceptibility.
Introduction
W
Because the number of SNP is very large with one SNP per 100-300 base pairs and a total number of 300-1000 million (Sachidanandam et al., 2001), development of an accurate, fast, and cost-effective method for detection of SNP is very necessary. At present, many SNP detection methods have been developed, such as allele-specific amplification (Wu et al., 1989; Ye et al., 1992), pyrosequencing (Alderborn et al., 2000; Fakhrai-Rad et al., 2002), rolling circle amplification (Qi et al., 2001; Li et al., 2010), and allele-specific hybridization (Guo et al., 1997; Howell et al., 1999). Most of these methods are based on polymerase chain reaction (PCR) technology. Usually, only one SNP can be detected at a time by using these methods, but some of them can be used to simultaneously determine multiple SNPs by employing multiplex PCR amplification (Colinas et al., 2000; Pourmand et al., 2002; Wang et al., 2006).
PCR, since its inception in 1985, with the advantages of being simple, rapid, and specific, has been widely used in various biological fields. Typically, genomic DNA extracted from peripheral blood is used as template for PCR amplification. However, as a multiple-step process is involved in the extraction of genomic DNA, it is time-consuming, labor-intensive, and prone to cause cross-contamination between samples, especially when a large number of samples are analyzed. Thus, many efforts have been made to develop a direct PCR amplification from whole blood without DNA extraction. Kogan et al. (1987) firstly realized this by using the supernatant of boiling blood sample for PCR amplification. After then, a variety of methods have been reported to be effective for direct PCR amplification from whole blood (Mercier et al., 1990; McCusker et al., 1992; Ohhara et al., 1994; Bu et al., 2008). Further, direct PCR from whole blood was also successfully used in PCR-based genotyping methods, including PCR restriction-fragment-length polymorphism, allele-specific PCR, SNaPshot mini-sequencing, and multiplex tetra-primer amplification refractory mutation system PCR (Kim et al., 2010). However, the compositions of these PCR solutions have not been disclosed.
It was found that some inhibitors, for instance, hemoglobin, immunoglobulin G, and anticoagulant agents existing in blood samples, can suppress PCR amplification (Abu Al-Soud and Radstrom, 2000). These inhibitors prevent the combination of DNA polymerase with the DNA template by interacting with genomic DNA and reduce the catalytic activity of DNA polymerase, resulting in inhibition of PCR amplification. Therefore, it might be possible to enable direct PCR amplification from whole blood by suppressing the electrostatic interaction between these inhibitors and genomic DNA. Since the genomic DNA is negatively charged in the conventional PCR system, this kind of interaction can be weakened when the surfaces of the inhibitors are also negatively charged. As we know, the net charge of protein depends on the pH value of solution. Thus, we can improve the pH value of PCR buffer to make the net charge of the protein inhibitor to be negative, so as to weaken the interaction between the protein inhibitors and genomic DNA, thereby eliminating the inhibitory effect of the protein inhibitors on PCR amplification. Abu Al-Soud and Radstrom (2000) found that the efficiency of PCR amplification can be improved by increasing the concentration of magnesium ion, which contributes to the maintenance of DNA polymerase activity and reduced the effect of anticoagulants, such as heparin and ethylenediaminetetraacetic acid (EDTA), in whole blood on PCR amplification. They also found that glycerol can enhance the efficiency of PCR amplification from whole blood. Consequently, in order to establish direct PCR amplification from whole blood, we assumed three measures to eliminate the suppression of these inhibitors: (1) increasing the pH value of the PCR solution to weaken the interaction between protein inhibitors and genomic DNA; (2) increasing the concentration of magnesium ions to reduce the impact of anticoagulants on PCR amplification; and (3) adding glycerol to improve the efficiency of PCR amplification.
Gastrointestinal cancers, including esophageal cancer, gastric cancer, and colorectal cancer, are the most common human malignancies. Numerous studies have showed significant association of SNPs with the development of these cancers (Dong et al., 2008). In this study, multiplex allele-specific amplifications from whole blood were successfully developed to simultaneously detect multiple SNPs and were used to detect 14 polymorphisms related to gastrointestinal cancers; this will help to achieve a rapid and low-cost genotyping method and can be used in large-scale association studies.
Materials and Methods
Blood samples
Our study was approved by the Institutional Ethics Review Board of Soochow University. A total of 238 unrelated Mainland Chinese outpatients, including 141 noncancer controls and 97 patients with gastric cancer, participated in this research after giving written informed consent. The patients were histologically confirmed to have gastric cancer by two pathologists. None of them had undergone radiotherapy or chemotherapy before surgery. Those with previous cancer or metastasizing cancer from other origins were excluded. The controls were matched with the gastric cancer patients by age (62.0±16.5 years for controls and 63.3±12.4 years for patients) and sex. All controls had no gastric disorders or personal or familial history of cancers, which was traced back to more than three generations and laterally to second- and third-degree relatives. Blood samples were collected using standard blood-draw procedures and stored in Vacutainer® tubes (BD) containing one of three commonly used anticoagulants to prevent clotting: 15 IU/mL heparin, 12.9 mM sodium citrate, and 4.5 mM EDTA. The blood samples were then stored at +4°C before use.
Single-plex allele-specific amplification of the rs9344 locus
To develop direct PCR amplification from whole blood, a single-plex allele-specific amplification of rs9344 locus was selected as an example for optimization of PCR conditions. The PCR was carried out in 25 μL containing 65 mM Tris, 50 mM potassium chloride (KCl), 2.0 mM magnesium chloride (MgCl2), 10% glycerol, 0.2 mM of each dNTP, 1 μL of whole blood, 0.4 μM of allele-specific primer and sequence-specific primer (Table 1), and 0.625 U of Taq DNA polymerase (MBI). Each PCR was carried out as follows: 94°C for 10 min, followed by 40 cycles of 94°C for 20 s, 50°C for 30 s, and 72°C for 45 s. Final extension was completed at 72°C for 7 min. Finally, 3 μL of PCR products was analyzed at room temperature by electrophoresis on a 2% agarose (LP0028A; Oxoid) gel containing 0.5 mg/mL ethidium bromide at 100 V for 20 min in 1×Tris-acetate-ethylenediaminetetraacetic acid buffer. Images of the gels were taken using GeneGenius BioImaging Systems (SynGene). Genomic DNA extracted from whole blood according to the phenol/chloroform method was also amplified under the same conditions as a positive control, while the negative control reaction included everything except for the template. All the oligonucleotides were synthesized and purified by GeneScript.
The uppercase letters (in bold) in the 3′-terminus of the primers represent the base specific to the allele type, and the lowercase letters represent the artificially mismatched base.
EC, GC, and CRC represent esophageal cancer, gastric cancer, and colorectal cancer, respectively.
SNP, single-nucleotide polymorphism.
Multiplex allele-specific amplification from whole blood
Multiplex allele-specific amplification on each blood sample was carried out in two separate tubes in 25 μL containing 65 mM Tris, 50 mM KCl, 2.0 mM MgCl2, 10% glycerol, 0.2 mM of each dNTP, 1 μL of whole blood, 0.4 μM of each sequence-specific primer, 0.4 μM of each allele-specific primer (Table 1), and 0.75 U of Taq DNA polymerase (MBI). The PCR cycling was carried out as follows: 94°C for 10 min, followed by 40 cycles of 94°C for 20 s, 50°C for 30 s, and 72°C for 45 s. Final extension was completed at 72°C for 7 min. Finally, 3 μL of allele-specific PCR products was analyzed by 2% agarose gel electrophoresis and then were imaged using GeneGenius BioImaging Systems.
Statistical analysis
Hardy-Weinberg equilibrium of alleles at individual loci was assessed by χ2 statistics. The differences of genotype and allele distribution between patients and controls were tested by Fisher's exact test and χ2 test. Odds ratios (ORs) with 95% confidence intervals and unconditional logistic regression models controlling for the effects of possible confounders were also computed. All of the statistical analyses were carried out with SPSS v11.5, and were done by two persons independently in a blind fashion. For all tests, p<0.05 was considered statistically significant.
Results and Discussion
Optimization of the PCR conditions
Since Tris base has the most significant effect on the pH value of the PCR solution, we first increased the pH value of the PCR solution by increasing the concentration of Tris base. Then, we optimized the concentrations of MgCl2 and glycerol. As shown in Figure 1A, the target DNA fragments could be successfully amplified with 25-100 mM of Tris base (lanes 3-6 in Fig. 1A), 1.5-2.5 mM of MgCl2 (lanes 13-15 in Fig. 1A), and 10%-12% of glycerol (lanes 24-25 in Fig. 1A). Furthermore under the conditions of 50 mM Tris base (lane 4 in Fig. 1A), 2 mM MgCl2 (lane 14 in Fig. 1A), and 10% glycerol (lane 24 in Fig. 1A), the amplifications were the most efficient. Thus, we used these conditions for direct PCR amplification from whole blood. As positive controls, we used purified genomic DNA for PCR amplification with various concentrations of Tris base, MgCl2, and glycerol. Successful amplifications from genomic DNA were observed under each of the conditions for successful amplifications from whole blood (lanes 2-7, 12-18, and 21-27 in Fig. 1B). In addition, the concentration ranges of Tris base, MgCl2, and glycerol for successful amplifications from genomic DNA are wider than that from whole blood (Fig. 1B). This was possibly due to the PCR inhibitors that occurred in the whole blood. However, our findings suggest that the PCR inhibition from whole blood can be overcome by increasing the concentrations of Tris base, MgCl2, and glycerol.
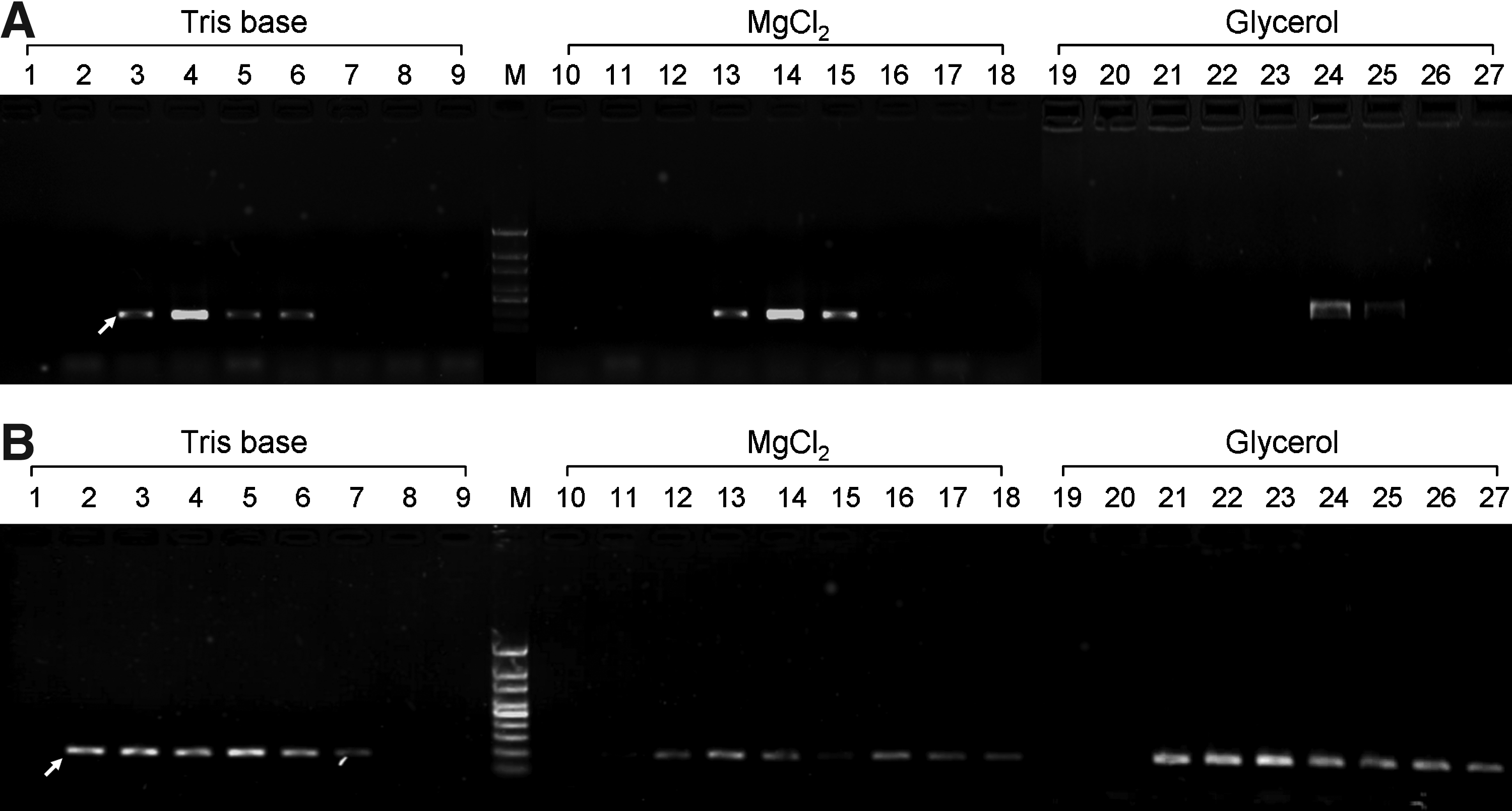
The effects of the concentrations of Tris base, MgCl2, and glycerol on the PCR amplification. The whole blood
Effect of anticoagulants on PCR amplification
As blood samples are usually stored in an anticoagulation tube before use, to study the impact of anticoagulants on PCR amplification, three commonly used anticoagulants, including heparin, sodium citrate, or EDTA, were added in the blood samples and were kept at +4°C for conservation. The results of PCR amplification showed that all of the whole blood samples containing heparin were successfully amplified, while the samples containing sodium citrate or EDTA were successfully amplified sometimes, but about 30% of the samples failed to be amplified (Fig. 2). Therefore, we used heparin as the default for treating blood samples to ensure direct PCR amplification from whole blood.
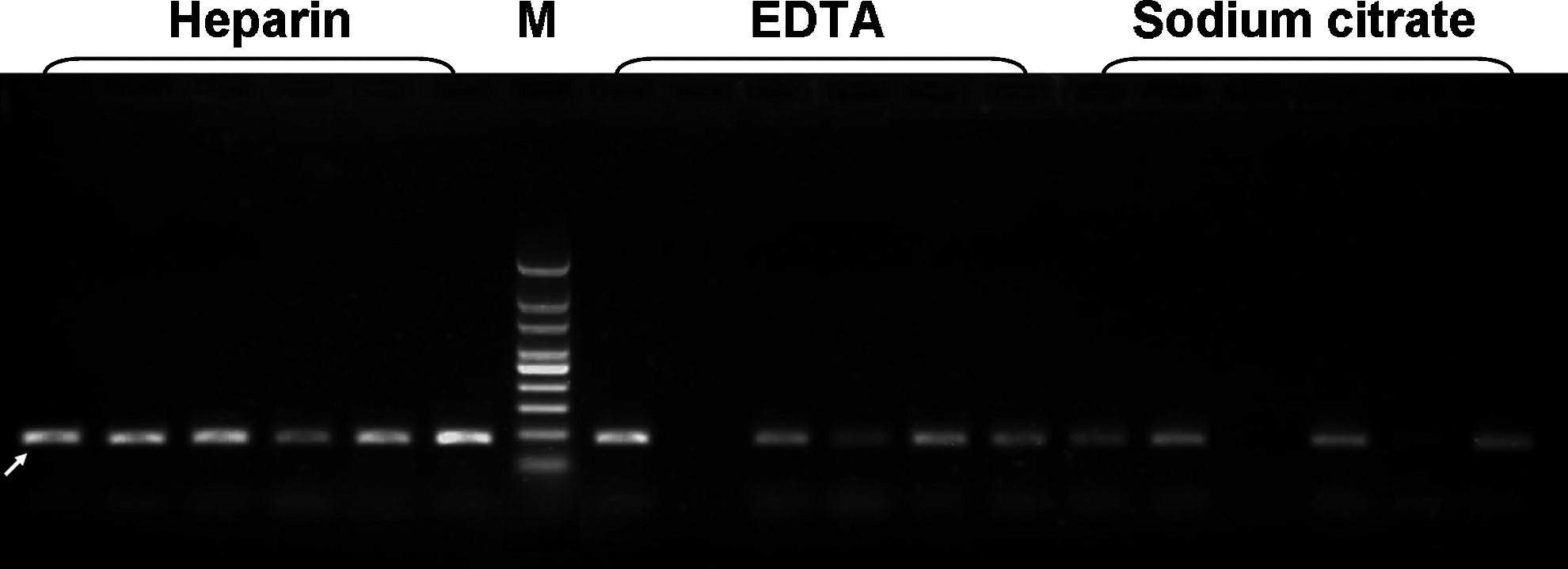
The effects of the anticoagulants of heparin, EDTA, and sodium citrate on the PCR amplification. Six whole blood samples each containing anticoagulants of heparin, EDTA, and sodium citrate were amplified. M represents DNA markers. The arrow indicates the specific amplicon. EDTA, ethylenediaminetetraacetic acid.
Multiplex genotyping
Under the optimized conditions, we performed multiplex allele-specific amplifications using allele-specific primers and sequence-specific primers in pairs of PCR tube for detecting multiple polymorphisms simultaneously. We established three-plex, five-plex, and eight-plex allele-specific amplifications to determine three polymorphisms associated with esophageal cancer, five polymorphisms associated with gastric cancer, and eight polymorphisms associated with colorectal cancer in 97 gastric cancer patients and 141 health controls, respectively. The typing results of four typical samples are shown in Figure 3. Based on the bands in a pair of electrophoretograms, it is easy to determine the genotypes of each SNP. To illustrate the effect of the proposed PCR buffer on the specificity and sensitivity of multiplex PCR, we performed the three-plex, five-plex, and eight-plex allele-specific amplifications from purified genomic DNA. The amplification results are shown in Figure 3D. These results indicated that the PCR buffer developed in this study was effective for multiplex allele-specific amplification from both the whole blood and genomic DNA samples, although weak small-size amplicons were observed in eight-plex allele-specific amplifications from the whole blood samples (Fig. 3C). To test the repeatability of the assays developed in this study, 30 randomly selected whole blood samples were detected by using these assays for three times each in a day. The typing results for each individual were exactly the same, indicating a good repeatability. To evaluate the reliability of these assays, the genomic DNAs were extracted from the 30 randomly selected whole blood samples, and were re-genotyped by Sanger's sequencing method (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/gtmb). The typing results for each individual were completely consistent with those obtained in this study, indicating a good reliability.
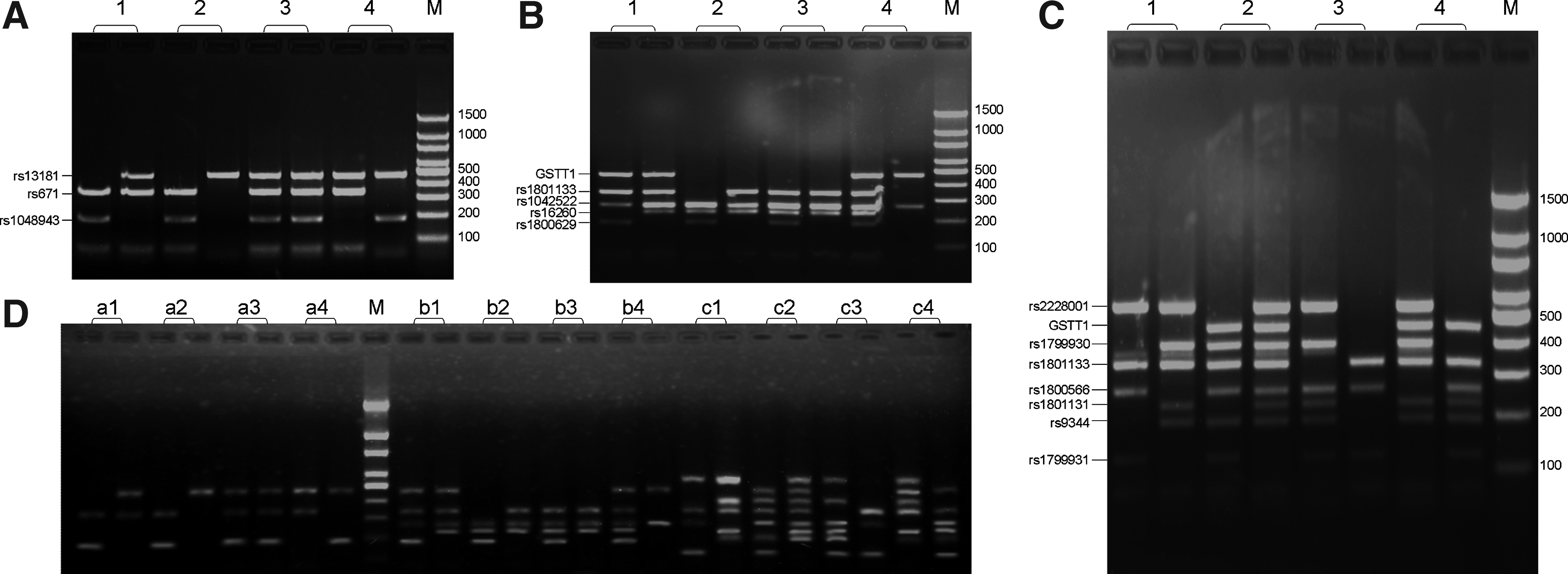
The electrophoretograms of three-plex
The associations of the polymorphisms with the risk of gastric cancer
In both control and patient populations, the alleles at the individual loci were in Hardy-Weinberg equilibrium with nonsignificant χ2 values (p>0.05), except for the rs1800629 locus in the patient population (χ2=46.46, p=8.16×10−11). Based on the typing results, the association of the 14 polymorphisms with the risk of gastric cancer in Chinese subjects was investigated. Comparison of genotype frequencies of rs1799931, rs9344, and rs1800629 showed significant differences between gastric cancer patients and controls (χ2=25.60, 17.12, and 27.69, respectively; p=2.77×10−6, 1.92×10−4, and 9.69×10−7, respectively; Table 2). No significant differences in genotype frequencies of the other SNPs were observed between patients and controls.
W and M represent the alleles before “>” and after “>” described in the third column of Table 1, respectively. The WW genotype was used as reference for χ2 test.
The W allele carrier was used as reference for χ2 test.
The W allele was used as reference for χ2 test.
P/C represents the number of gastric cancer patients versus the number of controls.
OR, odds ratio; 95% CI, 95% confidence interval.
In comparison with the SNP rs1799931 GG homozygotes, the GA heterozygotes and the AA homozygotes were associated with a significantly increased risk of gastric cancer with OR of 3.07 (p=9.98×10−5) and 11.4 (p=7.39×10−5), respectively (Table 2). The SNP rs1800629 AA homozygotes were also associated with a significantly increased risk of gastric cancer with OR of 20.2 (p=1.24×10−7) in comparison with the GG homozygotes (Table 2). Compared with the AA homozygotes of SNP rs9344, the AG (OR=0.51, p=0.035) and GG (OR=0.21, p=4.21×10−5) genotypes were related to obviously decreased risk of gastric cancer (Table 2). In addition, we looked for the correlation of an allele to the risk of gastric cancer and observed that rs9344 G allele (OR=0.43, p=1.54×10−5), rs1799931 A allele (OR=2.71, p=1.63×10−6), and rs1800629 A allele (OR=5.05, p=1.20×10−9) were significantly associated with the risk of gastric cancer (Table 2). Carriers of rs9344 G allele seem to be protected against gastric cancer at OR of 0.56 (p=0.011), and carriers of rs1799931 A allele (OR=2.02, p=2.88×10−3) as well as rs1800629 A allele (OR=3.35, p=2.19×10−4) are more likely to have gastric cancer. No association with gastric cancer was found for variants of rs671, rs13181, rs16260, rs1042522, rs1048943, rs1799930, rs1800566, rs1801131, rs1801133, rs2228001, and GSTT1 null/present. These results were consistent with those obtained in our previous study (Cao et al., 2011).
Conclusions
In this study, we developed multiplex allele-specific amplifications from whole blood directly for simultaneous determination of multiple genetic variants. Since the whole blood was used as starting material for PCR amplification and no special reagents and instruments were used in this method, the experimental cost, time consumption, and labor are very low. Moreover, as multiple genetic variants can be determined simultaneously and only two steps including allele-specific amplification and electrophoresis are involved in this method, the cost, time consumption, and labor for determination of each genetic variant were further reduced. The main limitation of this method is a relatively low throughput compared with chip-based platforms, such as the Affymetrix Gene Chip System (Fan et al., 2000) and the Illumina Golden Gate Assay (Gunderson et al., 2005). However, the throughput of this method can be greatly increased by the 96-well or 384-well detection platform and microarray gel electrophoresis (Wang et al., 2010) or microarray chip electrophoresis (Wang et al., 2007). By using this method, we detected the genotypes of 14 genetic variants and analyzed the associations of them with the risk of gastric cancer. We found that SNPs rs9344, rs1799931, and rs1800629 were significantly related to the occurrence of gastric cancer. These findings provided evidence that this method is accurate, fast, low cost, easy-to-do, and can be used in disease susceptibility and drug-metabolism-related studies.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 30901360 and No. 81270031), the National Science Foundation for Post-doctoral Scientists of China (No. 20080441077), the Jiangsu Postdoctoral Grant (No. 0802026C), and the Hui-Chun Chin and Tsung-Dao Lee Chinese Undergraduate Research Endowment.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.