
Abstract
Background: Rapid aneuploidy detection (RAD) methods constitute important complements to karyotyping in prenatal diagnosis. We evaluated the effectiveness of a method called high-resolution melting analysis of segmental duplications (SD-HRM) to serve as an alternative RAD method in prenatal diagnosis of common numerical chromosomal abnormalities (NCAs). Methods: We designed eight primary SD-HRM assays for the detection of chromosomes 13, 18, 21, X, and Y; 50 chorionic villus, 1105 amniotic fluid, and 395 cord blood samples were examined using these eight assays. For diagnosing samples that could not be diagnosed using primary assays, additional assays were designed for each target chromosome. Results: The success rate of eight primary SD-HRM assays ranged from 99.7% to 100%. For the distinguishable analyses, these eight assays attained high diagnostic sensitivities (100%) and specificities (99.9-100%). We differentiated 53 cases of NCAs from 1550 clinical samples; subsequent reference tests revealed that these assays attained 100% clinical sensitivity and specificity. The mosaic ratio of a 45,X/46,XX sample was also precisely calculated. Conclusions: The SD-HRM method was able to effectively detect common NCAs in 1550 prenatal samples. We propose that SD-HRM could serve as an effective alternative option to the currently used prenatal RAD methods.
Introduction
K
There is still no consensus on whether such rapid aneuploidy detection (RAD) methods should be used as standalone approaches for prenatal chromosomal testing (Caine et al., 2005; Boormans et al., 2009; de Jong et al., 2009; Ogilvie et al., 2009; Hills et al., 2010; Gekas et al., 2011). Opponents of RAD methods suggest that these assays may miss chromosomal abnormalities other than targeted aneuploidies, and those which result from RAD screening should be followed up with karyotyping (Test and Technology Transfer Committee, 2000; Caine et al., 2005; de Jong et al., 2009). However, proponents argue that chromosomal abnormalities with adverse outcomes missed by RAD are rare, because prenatal chromosomal diagnosis is mainly offered to pregnant women at a high risk who are selected by prenatal screening programs targeting trisomies 21 and 18 (Boormans et al., 2009; Ogilvie et al., 2009; Gekas et al., 2011).
However, this debate has not hampered the widespread use of well-recognized RAD methods, and, indeed, many new RAD methods have also emerged in recent years (Vialard et al., 2011; Yan et al., 2011; Dan et al., 2012; Guo et al., 2012). Technically, each RAD method possesses both advantages and limitations. Therefore, the choice of specific RAD methods for prenatal testing is strongly dependent on the demands of each case and the available resources.
In previous studies, we described a novel one-step RAD method called high-resolution melting analysis of segmental duplications (SD-HRM) (Guo et al., 2012). It was shown that SD-HRM could accurately differentiate samples of patients with common aneuploidies from those of unaffected controls, while markedly simplifying assay setup and analysis and reducing time and costs compared with existing RAD methods. In the present study, we have applied SD-HRM to the detection of common NCAs from 1550 clinical samples to evaluate its effectiveness as an alternative option for rapid prenatal diagnosis.
Materials and Methods
Ethics statement
Written informed consent was obtained from each participant before sample collection, and the study protocol was approved by the Research Ethics Committee of Xiamen Maternal and Child Health Hospital.
Subject enrollment
Chorionic villus, amniotic fluid, and cord blood are harvested corresponding to different gestational age. For amniotic fluid and cord blood sample collection, pregnant women at a risk for fetal aneuploidy were recruited at the Prenatal Diagnosis Center of Xiamen Maternal and Child Health Hospital during the period from November 2011 to December 2012. For SD-HRM, 10 mL of amniotic fluid was collected from 1105 participants at the gestational age of 16-22 weeks, and 1 mL of cord blood was collected from 395 participants at the gestational age of 22-26 weeks. The karyotypes of all samples were subsequently confirmed via karyotyping.
Considering that the amount of remaining chorionic villi were too low for SD-HRM analysis after accounting for requirements of prenatal karyotype analysis, we instead collected 50 chorionic villus samples from first-trimester (9-14 weeks) spontaneous abortions. The SD-HRM results were validated using fluorescence in situ hybridization (FISH) assays.
Sample pretreatment and DNA extraction
For pretreatment of chorionic villus samples, maternal decidua, mucus, and other contaminants were removed with the aid of a microscope. 10-15 mg of chorionic villi was then homogenized in 200 μL of sterilized normal saline solution using a homogenizer (IKA). For pretreatment of amniotic fluid samples, 10 mL of amniotic fluid was transferred to microcentrifuge tubes and centrifuged at 16,000 g for 5 min to concentrate the fetal cells. Fetal cells were then re-suspended in 200 μL of sterilized normal saline solution. For pretreatment of cord blood samples, 100 μL of EDTA-anticoagulated cord blood was mixed with 100 μL of sterilized normal saline solution for a final volume of 200 μL.
Genomic DNA was extracted from the pretreated samples using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer's protocol. An additional 75 μg of RNase A (BBI) was added and co-incubated during the lysis step when cord blood samples were processed. Extracted DNA was dissolved in 80 μL of AE buffer (Qiagen), and its concentration was determined by measuring the ultraviolet absorbance at 260 nm with the NanoVue Plus spectrophotometer (GE Healthcare).
PCR and HRM
PCR amplification and HRM analysis were performed on a LightCycler 480 II thermocycler (Roche Applied Science) in 20 μL reactions containing 1×LightCycler 480 High-Resolution Melting Master mix (Roche), 2.5 mM Mg2+, and 250 nM of each forward and reverse primer (Invitrogen). Generally, 25 ng of genomic DNA was used as the template. For DNA samples with concentrations lower than 5 ng/μL, 5 μL of these samples were used as the PCR template. The PCR cycling conditions were as follows: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, 60°C for 15 s, and 72°C for 15 s. High-resolution melting began with denaturation at 95°C for 1 min and renaturation at 40°C for 1 min, followed by melting from 60°C to 90°C at a ramp rate of 0.03°C/s and the acquiring of fluorescence data at a frequency of 20 readings/°C.
Data analysis
High-resolution melting data were analyzed as graphs of fluorescence versus temperature with the aid of the Gene Scanning Software (version 1.5.0; Roche). The intact melting curve analysis comprised four steps: (a) Data were normalized by selecting the linear regions of melting curves before and after DNA dissociation; (b) samples were grouped by shifting the temperature axes of the normalized melting curves; (c) the relative signal differences were plotted versus temperature after the samples were clustered into groups with the aid of the selected reference melting curve; and (d) the relative signal difference between groups at a given temperature was statistically analyzed. Statistical analyses were performed using OriginPro 7.5 software (OriginLab Corporation).
In the clinical application, we tested 15 to 25 samples once per sampling cycle. For each assay, the corresponding aneuploidy and unaffected samples were simultaneously analyzed as controls. For example, for the 13vs18 assay, a trisomy 13, a trisomy 18, and an unaffected sample were used as controls. Consequently, with the aid of control samples, we could classify the clinical samples at the melting curve analysis step (c) based on the relative signal difference. Step (d) was performed only when some parameters, such as analytical specificity, were evaluated in depth.
The influence of DNA template amount on analytical specificity
Three assays were randomly selected for this part of the study: 13vs18, 12vs21, and 2vsX assays. For each assay, two panels of DNA samples (n=10, respectively) with different karyotypes were analyzed. Analytical specificities between panels were subsequently evaluated using 5, 10, 25, or 50 ng of DNA template. F value (one-way ANOVA) was used as the parameters for measuring analytical specificity. Generally, the F value increases with increases in the difference between the two groups.
The influence of RNA on analytical specificity
Eight chorionic villus samples, eight cord blood samples, and 10 amniotic fluid samples were collected. Samples were divided into two for DNA extraction, resulting in two panels of 26 samples, each consisting of eight chorionic villus, eight cord blood, and 10 amniotic fluid samples. One panel of samples was processed without involving the RNase treatment, and the other was treated with 75 μg of RNase A during the lysis step. The extracted nucleic acid was then quantified according to the OD260 value, and 25 ng of nucleic acid was analyzed using the SD-HRM assays, 13vs18 and 12vs21.
Standard deviations of melting curves within panels and the difference in the mean value (Δmean) between panels were used as the parameters for measuring analytical specificity. Generally, analytical specificity exhibits a negative correlation with the standard deviation and Δmean. Using data analysis step (d), the standard deviations of melting curves corresponding to the two panels were calculated and compared with each other; three types of samples from each panel were analyzed separately.
Results
Study design
The flowchart used for this study is illustrated in Figure 1. Based on our previous work (Guo et al., 2012), we selected eight pairs of segmental duplicates and redesigned primers within them. Using this strategy, dosages of chromosomes 13, 18, and 21, and the sex chromosomes can be dually confirmed by these eight assays. To evaluate the sensitivity and specificity of SD-HRM, karyotyping or FISH was performed in parallel for each sample.
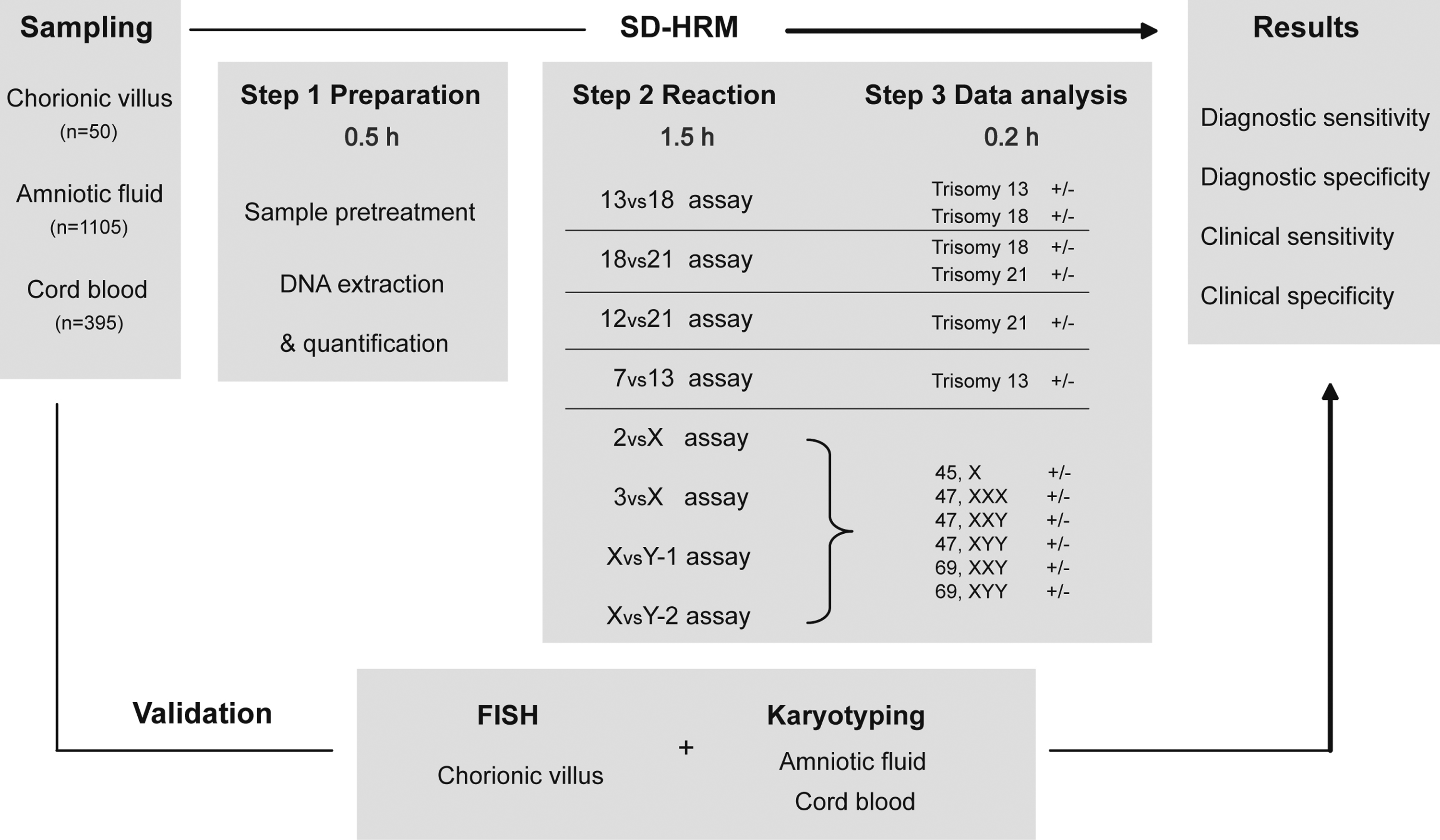
Flowchart of prenatal diagnosis of trisomies 13, 18, and 21, and sex chromosome aneuploidies using high-resolution melting analysis of segmental duplications (SD-HRM). Chorionic villus, amniotic fluid, and cord blood samples were collected. After sample pretreatment, DNA was prepared from the corresponding clinical samples and analyzed using the eight primary assays. The dosages of chromosomes 13, 18, and 21, and the sex chromosomes could be dually confirmed using these eight assays. The sensitivity and specificity of SD-HRM were evaluated by examining the results of karyotyping or FISH carried out in parallel on the same samples. The turnaround time from DNA extraction to result was less than 2.5 h using SD-HRM.
The typical melting profiles of eight primary assays are illustrated in Figure 2. For cases in which potential discrepancies between two assays for a corresponding chromosome were observed, we also designed one additional primer set for each target chromosome. In these instances, results were assessed using these primers as “additional assays.” Primer and amplicon information is available in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/gtmb).

Melting curve profiles of corresponding controls for eight primary SD-HRM assays. The corresponding aneuploidy and unaffected samples were simultaneously analyzed as controls with clinical samples for each assay. Consequently, we could classify the clinical samples by comparing the melting curve profiles with those of controls.
Considering that the amount of fetal cells which can be separated from 10 mL of amniotic fluid is limited, we first evaluated the influence of DNA template amount on analytical specificities of SD-HRM assays (Materials and Methods section). As shown in Figure 3A and 3B, 25 ng was the optimal DNA template amount, affording the highest analytical specificity, and it was, therefore, used throughout this study.
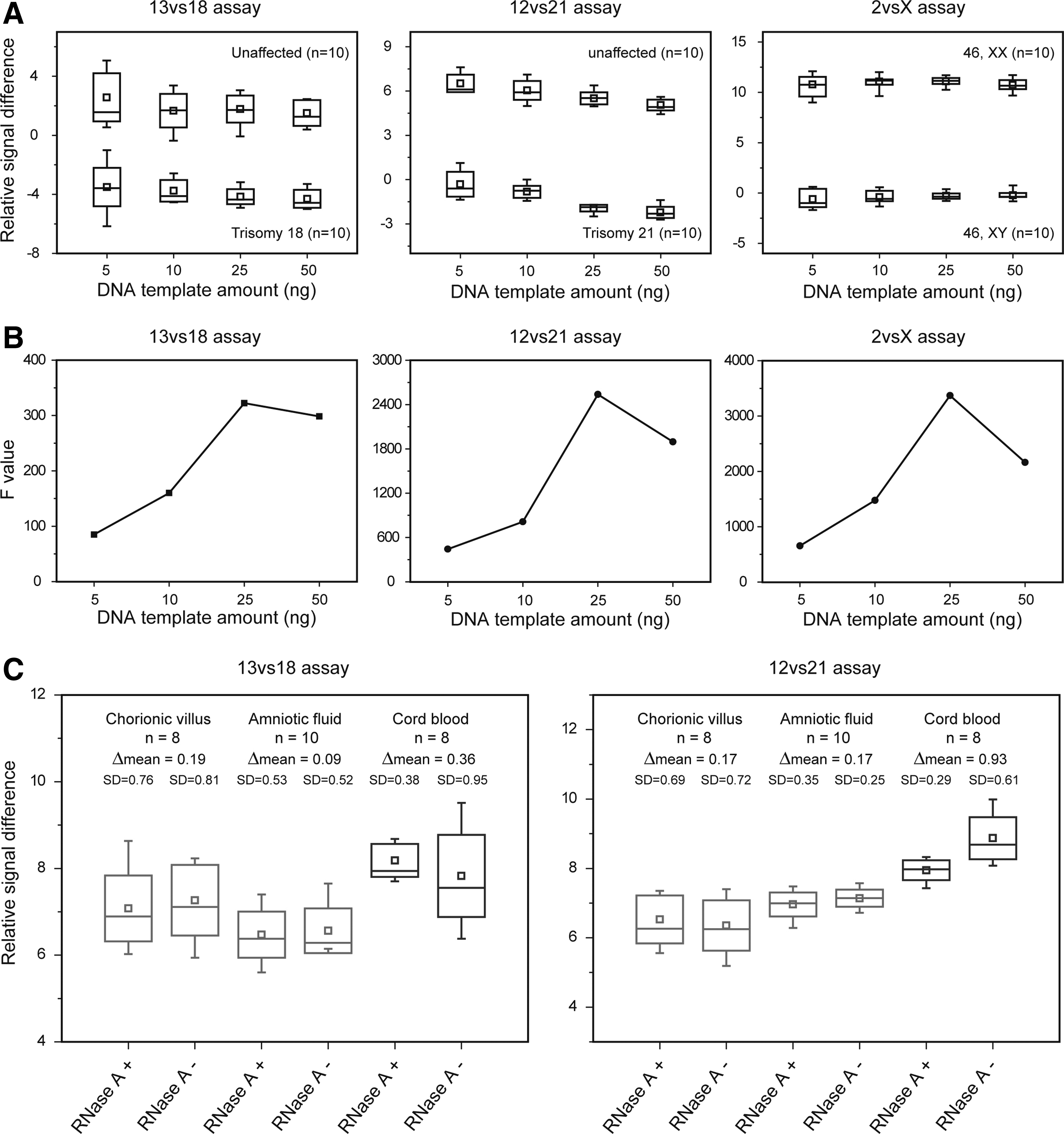
Analytical specificities of SD-HRM assays. Relative signal differences at 81.0°C (13vs18 assay), 75.5°C (12vs21 assay), and 78.6°C (2vsX assay) were extracted for statistical analysis.
RNA in cord blood decreases the analytical specificity
In comparison with chorionic villus and amniotic fluid samples, RNA was found to be more abundant in cord blood samples (Supplementary Fig. S1). Therefore, we also evaluated the influence of residual RNA on analytical specificities of SD-HRM assays (Materials and Methods section). As shown in Figure 3C, in the cord blood samples, the standard deviation of melting curves was much larger in the RNase-untreated panel than that of the RNase-treated panel for both 13vs18 and 12vs21 assays. Unlike cord blood samples, such a finding was absent in chorionic villus and amniotic fluid samples. Moreover, the Δmean between the two panels was much larger in cord blood samples than in the other sample types. These results indicated that abundant RNA in cord blood samples decreased the analytical specificity of SD-HRM assays.
Mosaicism detection
A 45,X/46,XX mosaicism was detected using the 2vsX and 3vsX assays. To measure the mosaic ratio of this sample, we prepared serial mixed samples containing a different ratio of 45,X DNA by diluting 45,X DNA with 46,XX DNA. The mixed samples were then analyzed with the 2vsX and 3vsX assays to establish standard quantification curves (Fig. 4). Using this approach, we found that the mosaic ratio of mosaicism was 53.3% (51.9-54.7%, 95% confidence interval [CI]) with the 2vsX assay and 58.5% (52.5-64.1%, 95% CI) with the 3vsX assay (Fig. 4B). The mosaic ratio was 54%, as calculated from subsequent karyotype analyses (45,X[27]/46,XX[23]). Based on a 95% confidence level, a mosaic ratio of 54% falls within both confidence intervals attained from 2vsX and 3vsX assays, thus revealing high concordance between karyotyping and SD-HRM.

Quantification analysis of the mosaic ratio of mosaicism. Each sample was analyzed in triplicate.
Sensitivity and specificity
As shown in Table 1, all 1550 prenatal samples were successfully distinguishable by 18vs21, 12vs21, 7vs13, 3vsX, and XvsY-1 assays. We found that 4, 1, and 2 samples could not be analyzed using the 13vs18, 2vsX, and XvsY-2 assays, respectively. The melting curve profiles of indistinguishable samples substantially deviated from those of controls; these samples were then successfully classified using additional assays (Supplementary Figs. S2 and S3). Sequencing results from these samples revealed the existence of point mutations (Supplementary Fig. S4). For the distinguishable analyses, the results of seven primary SD-HRM assays were fully concordant with either karyotyping or FISH, thus attaining 100% diagnostic sensitivity and specificity (Table 1). The chromosome 18 dosage of 1 sample was controversial between the results determined from 13vs18 and 18vs21 assays; however, the subsequent use of an additional assay confirmed the results of the 13vs18 assay (Supplementary Fig. S5). Thus, the 18vs21 assay attained 99.9% diagnostic specificity for the detection of trisomy 18 (Table 1). Using these eight primary SD-HRM assays in conjunction with the additional assays, we accurately detected 25 cases of trisomy 21, 11 cases of trisomy 18, 4 cases of trisomy 13, 7 cases of 45,X (one was a mosaicism), 1 case of 47,XXX, 1 case of 47,XXY, 1 case of 48,XYY,+18, and 3 cases of 69,XXY from 1550 prenatal samples, and attained 100% clinical sensitivity and specificity based on subsequent confirmation by karyotyping or FISH (Table 2).
One sample in 12 is a double trisomy. Its karyotype is 48, XYY, +18.
One sample in 7 is a 45, X mosaicism. Its karyotype is 45,X[27]/46,XX[23].
SD-HRM, high-resolution melting analysis of segmental duplications.
Discussion
In comparison with the most widely used prenatal RAD methods, for example, quantitative fluorescent PCR (QF-PCR) (Mansfield, 1993) and multiplex ligation-dependent probe amplification (MLPA) (Schouten et al., 2002), SD-HRM assays are less complex, less prone to carry-over contamination, take less time complete, and are less costly (Guo et al., 2012). In addition, SD-HRM is amenable to automation and provides sufficient capacity for high-throughput screening (Seipp et al., 2010), thus meeting the demands of large-scale testing.
Generally, chorionic villus, amniotic fluid, and cord blood are the most common materials harvested for prenatal diagnosis corresponding to different gestational age. Therefore, we examined all three sample types in this study. We found that the mean concentration of amniotic fluid-derived DNA samples was just 19.7 ng/μL due to the limited amount of fetal cells which could be separated from 10 mL of amniotic fluid, which is much lower than that of samples derived from chorionic villus and cord blood samples (Supplementary Fig. S6). However, the concentrations of 98.3% of amniotic fluid-derived DNA samples reach the level of 5 ng/μL, which means that 5 μL of these DNA samples would provide 25 ng of DNA template, which is optimal for SD-HRM assays to achieve the highest analytical specificity (Fig. 3B). Lower amounts of DNA template may result in unstable proportions of segmental duplicates due to the Poisson distribution, and they may decrease the analytical specificity. However, we did not observe any failed or false detections for the 1.7% of amniotic fluid-derived DNA samples for which yields were lower than the optimal amount.
We also found that RNase treatment increased the analytical specificity of SD-HRM assays when analyzing cord blood samples. Without RNase treatment, a considerable amount of RNA remained in the DNA solution extracted from cord blood samples compared with extractions from chorionic villus and amniotic fluid samples (Supplementary Fig. S1). Residual RNA markedly decreased the analytical specificities of SD-HRM assays (Fig. 3C). Two explanations may account for such findings. First, when DNA was quantified based on the OD260 value, the existence of RNA lowers the actual DNA concentration below the theoretical value (Supplementary Fig. S1); we have noted that less than 25 ng of DNA can result in a decrease of analytical specificity (Fig. 3B). Second, an overabundance of RNA may also result in deviations of the high-resolution melting curve profiles. Therefore, additional RNase treatment is highly recommended for DNA extraction from cord blood samples. In contrast, for chorionic villus and amniotic fluid samples, following the routine DNA extraction protocol without RNase treatment is sufficient (Fig. 3C and Supplementary Fig. S1).
As in our previous study, SD-HRM exhibited a remarkably high resolution, and was able to detect a change in chromosome dosage as low as 1.05-fold (Guo et al., 2012). This level of resolution could permit the detection of most targeted chromosomal mosaicism. In the present study, the capability of mosaicism detection was further validated by accurate quantification of the mosaic ratio of a 45,X/46,XX sample (Fig. 4).
Our analysis of 1550 clinical samples confirmed the previous hypothesis that the adoption of segmental duplicates as detection targets ensures the universality and accuracy of SD-HRM assays (Guo et al., 2012). In the present study, the universality of SD-HRM was revealed by the high success rate (99.7-100%) of the eight primary assays used (Table 1); the high accuracy of SD-HRM was revealed by the high diagnostic sensitivities (100%) and specificities (99.9-100%) of these assays and the 100% clinical sensitivity and specificity of SD-HRM for prenatal diagnosis of NCAs (Table 2). Although results at a given assay may vary slightly depending on ethnic background, the target segmental duplicates are easy to optimize. Point mutation and copy number variation (CNV) are two inevitable issues that could affect the universality and accuracy of SD-HRM. For example, we were not able to definitively analyze eight samples for point mutations and/or CNV at the target segmental duplicate loci; however, these eight samples could simply be validated using the additional assays (Supplementary Figs. S2, S3, and S5).
Theoretically, increasing the target density for each chromosome would increase confidence in abnormal calls compared with solely considering data from the two target loci we tested. However, by evaluating 1550 clinical samples, we found that for aneuploidy detection, both the clinical sensitivity and specificity of our method (100%) were equivalent to QF-PCR and MLPA, which possess higher target density (Boormans et al., 2010; Papoulidis et al., 2012). Such data implied that two target loci per chromosome were sufficient for ensuring precision in prenatal diagnosis. Furthermore, although a few additional assays may be required when only two target loci per chromosome are used, this strategy is more cost effective than screening three or more target loci per chromosome using this method. However, further evaluation in larger sample sizes would be required to validate these conclusions before this method is used as a standalone test.
SD-HRM also has limitations. Although its high resolution allows for the detection of maternal cell contamination (MCC) in male fetus samples by observing the deviation of melting profiles of XvsY assays, SD-HRM will miss MCC in female fetus samples. In this regard, there is a potential risk of misdiagnosis if MCC occurs in an abnormal female fetus. However, as previously shown (Guo et al., 2012), even in cases in which MCC occurrence is as high as 90%, evidence for possible fetal aneuploidies could still be observed from the melting profiles of corresponding SD-HRM assays. In such cases, the screening for MCC should be conducted to differentiate MCC from fetal mosaicism. It should also be noted that 69, XXX, is indistinguishable when using SD-HRM, because melting profiles for these samples are identical to those of normal females. However, given that triploid fetuses are often associated with ultrasound anomalies, the clinical consequences of this are likely to be insignificant.
In conclusion, SD-HRM exhibits high clinical sensitivity and specificity in the clinical application of prenatal RAD and reveals remarkably high resolution, facilitating accurate quantitative detection of mosaicisms. Prenatal RAD can be carried out using this technique in 2 h at a relatively low cost (less than $5 per sample, for reagents only); its one-step protocol minimizes labor and enables easy automation. In this context, we propose that SD-HRM could serve as an effective alternative to other currently used methods for the rapid prenatal diagnosis of common NCAs.
Footnotes
Acknowledgments
The authors thank the cytogenetic laboratory of Xiamen Maternal and Child Health Hospital for karyotype and FISH analysis; Li Sun and Yasong Xu of Xiamen Maternal and Child Health Hospital for sample collection; Wei Zhang of Florida Atlantic University for kindly providing support with statistics; and Zhiqiang Lin for running the experiment.
The authors are also grateful for the support rendered by Y. Zhou, Fujian Provincial Science and Technology Key Project (project no. 2012D066) and National Key Technology R&D Program in the 11th Five-year Plan of China (project no. 2006BAI05A10); Q. Guo, National Science Foundation for Young Scholars of China (project no. 81201361); and Foundation for Young Scholars of Fujian Provincial Department of Health Office (project no. 2010-2-111).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.