
Abstract
Background: Glutathione S-transferases (GSTs) play an important role in the detoxification of a wide variety of toxicants. Among GSTs, GSTM1 and GSTT1 polymorphic deletions and the GSTP1 Ile105Val polymorphism were often studied in combination in many diseases because of their additive effects, but they were usually genotyped by two separate methods. Aim: The purpose of the present study was to develop a simple and reliable method to simultaneously detect these three polymorphisms. Methods: The three polymorphisms of 259 volunteers were genotyped using a multiplex polymerase chain reaction (PCR) method based on a tetra-primer amplification refractory mutation system-PCR (T-ARMS-PCR), and the results were validated by multiplex PCR for GSTM1 and GSTT1 polymorphisms and DNA sequencing of the GSTP1 Ile105Val polymorphism, respectively. Results: The multiplex PCR method of GSTM1, GSTT1, and GSTP1 polymorphisms based on T-ARMS-PCR can simultaneous detect the three polymorphisms in a single PCR. The results of this method are in perfect accord with the results of the multiplex PCR method of determining GSTM1, GSTT1 polymorphisms, and DNA sequencing of GSTP1 Ile105Val polymorphism. Conclusion: The novel multiplex PCR method of GSTM1, GSTT1, and GSTP1 polymorphisms is simple, fast, low-cost, and reliable for the simultaneous detection of GSTM1, GSTT1, and GSTP1 Ile105Val polymorphisms.
Introduction
G
The tetra-primer amplification refractory mutation system-PCR (T-ARMS-PCR) method is a fast, cost-effective, reliable method for single-nucleotide polymorphism (SNP) genotyping (Ye et al., 2001). Because it does not require any special equipment, the assay can be set up in most molecular biology laboratories (Etlik et al., 2011). A quadruplex T-ARMS-PCR method has been developed to detect three SNPs and one indel polymorphism in a single PCR (Lajin et al. 2012a).
The aim of the present work was to develop a multiplex PCR method for the simultaneous detection of GSTM1, GSTT1, and GSTP1 polymorphisms in a single PCR to reduce detection cost and time.
Materials and Methods
Participants
The study participants consisted of 259 volunteers living in Jiangxi, China. Each participant provided informed consent. The study was approved by the Medical Ethics Committee of the First Affiliated Hospital of Nanchang University.
DNA extraction
For DNA extraction, 2 mL of blood was obtained from each participant. The genomic DNA was purified from whole blood using a DNA isolation kit (Bioteke, Beijing, China), and the procedure was performed according to the instruction manual.
Multiplex PCR method of the three polymorphisms
Table 1 shows the sequences of primers used for multiplex PCR of the three polymorphisms. A mismatch at the third nucleotide from the 3′ terminus of the GSTP1 allele-specific primers was introduced to maximize specificity. The primers for the GSTM1 and GSTT1 polymorphisms can be found elsewhere (Chen et al., 1996; Yadav et al., 2010). The PCR (10 μL) contained 5 μL of 2× Taq master mix (SinoBio, Shanghai, China), eight primers (the final concentration of each primer is listed in Table 1), and 10 ng of genomic DNA. PCR cycling was carried out on a MG96G thermal cycler (LongGene, Hangzhou, China). Amplification was performed by initial denaturation at 94°C for 3 min, followed by 35 cycles of 94°C for 20 s, 60°C for 20 s, and 72°C for 30 s. A final extension of 72°C for 5 min was performed and then the reaction was cooled to 10°C. PCR products were identified by electrophoresis on a 2% agarose gel. The gel was photographed using a JS-680B gel imaging system (Peiqing, Shanghai, China).
Specific nucleotides are underlined; specificity-enhancing mismatches are shown in bold italics.
Method validation
The β-globin gene forward (5′-CAACTTCATCCACGTTCACC-3′) and reverse (5′-GAAGAGCCAAGGACAGGTAC-3′) primers were used as an internal positive control for the multiplex PCR method to avoid false-negative results (Yadav et al., 2010). The PCR was performed in a 10-μL mixture containing 5 μL of 2× Taq master mix, 0.2 μM of each primer, and 10 ng of genomic DNA. Amplification was performed by initial denaturation at 94°C for 3 min, followed by 35 cycles of 94°C for 20 s, 60°C for 20 s, and 72°C for 25 s, and a final extension of 72°C for 5 min. The product lengths were 219 bp, 459 bp, and 268 bp for GSTM1, GSTT1, and β-globin, respectively. Absence and presence of PCR products for GSTM1 and GSTT1 were considered as null and positive genotypes, respectively.
For the GSTP1 Ile105Val polymorphism, 26 samples (about 10% of the total samples) were randomly sequenced (Sangon, Shanghai, China). The PCR (10 μL) contained 5 μL of 2× Taq master mix, 0.2 μM of GSTP1-common-F, 0.2 μM of GSTP1-common-R, and 10 ng of genomic DNA. The PCR cycling conditions were the same as those of the multiplex PCR method.
Results
Multiplex PCR of the three polymorphisms
A representative result of agarose gel electrophoresis is shown in Figure 1. The amplified DNA fragments are 284 bp for GSTP1 105Val (313G), 347 bp for GSTP1 105Ile (313A), 219 bp for GSTM1, and 459 bp for GSTT1, as well as a common 586 bp band for GSTP1. The bands were sufficiently clear for each sample to be genotyped correctly. All genotype combinations of the three polymorphisms are demonstrated in Fig. 1. Table 2 shows the observed genotype frequencies for GSTP1 Ile105Val, GSTM1, and GSTT1 among the 259 volunteers.

Genotyping of three polymorphisms by multiplex polymerase chain reaction. Lanes 1-11 show all genotype combinations of the three polymorphisms, and lane 12 is a negative control. Lane M is DNA marker I.
Multiplex PCR of GSTM1 and GSTT1
All samples were genotyped by the multiplex PCR method for GSTM1 and GSTT1. No difference in genotype assignment was observed in any of the samples. Figure 2 shows the electrophoretogram of the multiplex PCR for GSTM1 and GSTT1 of 11 different samples, corresponding to the samples described in Figure 1.
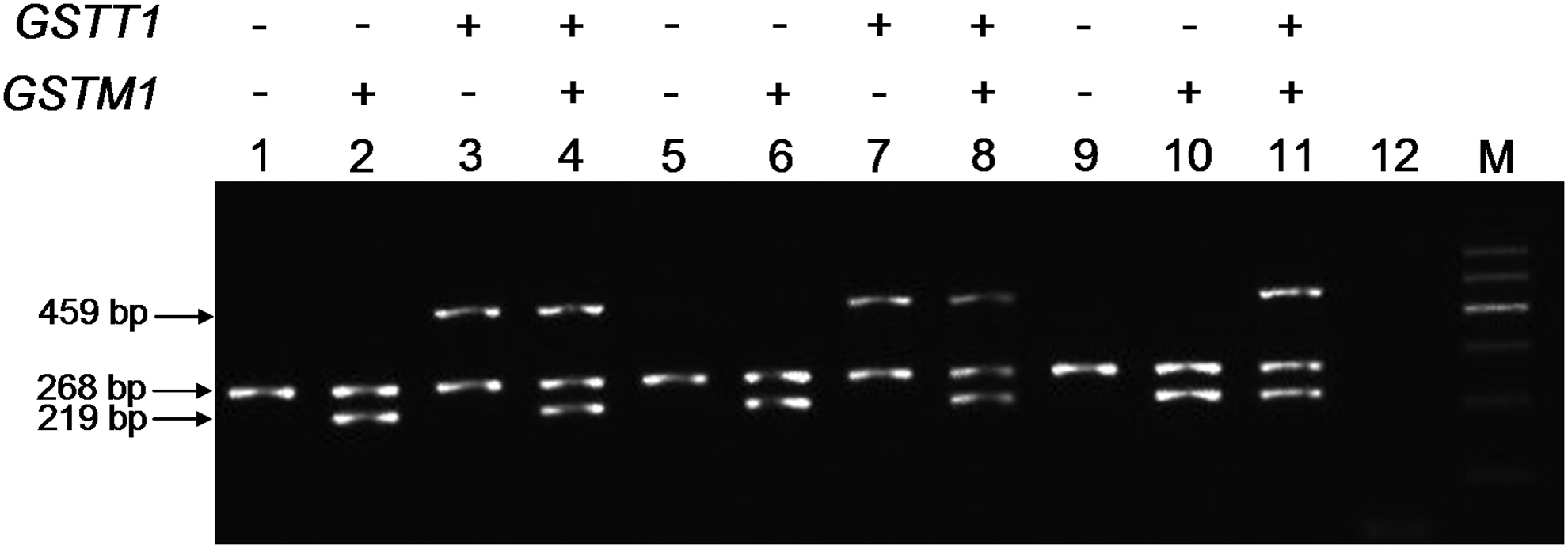
Genotyping of GSTM1 and GSTT1 by multiplex polymerase chain reaction. Lanes 1-11 correspond to the samples described in Figure 1, and lane 12 is a negative control. Lane M is DNA marker I.
DNA sequencing
The results of DNA sequencing for the GSTP1 Ile105Val polymorphism were consistent with the results of the multiplex PCR method for the three polymorphisms.
Discussion
GSTM1, GSTT1, and GSTP1 polymorphisms have been extensively investigated because they may influence enzyme activity. To date, many meta-analyses have investigated the relationships between GST polymorphisms and many different diseases. The dual null genotype of GSTM1 and GSTT1 was associated with increased risk of prostate cancer (Gong et al., 2012), diabetes mellitus (Zhang et al., 2013), and hepatocellular carcinoma in Chinese people (Liu et al., 2013). The GSTM1 null genotype was associated with increased risk of prostate cancer (Gong et al., 2012), childhood asthma ( Li et al., 2013), male infertility (Ying et al., 2013), diabetes mellitus (Zhang et al., 2013), and oral cancer in the Asian population (Zhang et al., 2011), while the GSTT1 null genotype was associated with increased risk of chronic myeloid leukemia (Zintzaras, 2009) and diabetes mellitus (Zhang et al., 2013). The GSTP1 Ile105Val polymorphism may increase susceptibility to bladder cancer (Wang et al., 2013) and breast cancer in the Asian population (Lu et al., 2011). The GSTT1 null genotype and GSTP1 Ile105Val polymorphism are also associated with high risk of prostate cancer (Gong et al., 2012), and the GSTM1 null genotype and GSTP1 Ile105Val polymorphism are associated with high risk of primary open-angle glaucoma (Yu et al., 2013). Compared with single gene analysis, the investigation of multiple genetic polymorphisms could contribute to a better understanding of the effects of susceptibility genes on cancer risk (Palma et al., 2010). Thus, it is necessary to develop a simple and reliable method to simultaneously detect these three polymorphisms.
Because of its simplicity and reliability, T-ARMS-PCR has been widely used to genotype SNPs and mutations (Etlik et al., 2011; Lajin et al., 2012a; Lajin et al., 2012b). We previously genotyped two SNPs in the CYP19A1 gene using the T-ARMS-PCR method (data not shown). In our experience, to achieve optimal results with T-ARMS-PCR in a short period, the following three steps should be taken. The first step is to find a heterozygous sample. By using an annealing temperature of 56°C, two allele-specific fragments, together with the control fragment, are amplified in 10 samples (the primer concentrations of specific fragments are twice those of control fragments). The sample will be heterozygous if the products for both of the allele-specific fragments are observed, whereas a product for only one specific fragment should emerge in other samples. The second step is determination of an optimal annealing temperature. For the determined heterozygous sample, the annealing temperature is altered from 56°C to 65°C, and the two specific fragments are amplified together with the control fragment. The temperature at which the three fragments are brightest is the optimal annealing temperature. The final step is to determine the optimal primer concentrations. T-ARMS-PCR is performed at the previously determined optimal annealing temperature with different primer concentrations. The primer concentrations at which the two specific fragments are evenly bright are considered optimal concentrations for T-ARMS-PCR. Consistent with the results of Lajin et al., we also found that introducing mismatches at the third nucleotide from the 3′ terminus of the primers played a crucial role in the specificity of T-ARMS-PCR, with allele-specific primers losing their specificity upon removal of these mismatches (Lajin et al., 2012b). Moreover, Taq master mix may be an important factor that affects band intensities and the absence or presence of the common band. To get uniform results, Taq master mix should be of the same production lot.
On the basis of T-ARMS-PCR of the GSTP1 Ile105Val polymorphism, we developed a multiplex PCR method for the simultaneous detection of GSTM1, GSTT1, and GSTP1 Ile105Val polymorphisms. For 10 samples, this method can be completed within 4 h after specimen receipt (60 min for DNA extraction, 100 min for PCR amplification, and 40 min for agarose gel electrophoresis). The results of this method are in accordance with the results of the multiplex PCR method for GSTM1 and GSTT1 and DNA sequencing of GSTP1. This confirmed the reliability of this method for the simultaneous detection of GSTM1, GSTT1, and GSTP1 polymorphisms. This method is more convenient and economical than the traditional PCR restriction fragment-length polymorphism because it does not require incubation with costly restriction enzymes for several hours. Other methods, such as denaturing high-performance liquid chromatography, real-time PCR, capillary electrophoresis, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry, and direct DNA sequencing, can also be used to detect these polymorphisms, but they require expensive equipment that might not be routinely available in most laboratories (Etlik et al., 2011). At present, we are using this method to investigate the relationships between diabetic retinopathy and GSTM1, GSTT1, and GSTP1 polymorphisms.
Conclusion
In summary, novel multiplex PCR is an accurate, simple, fast, low-cost method for the simultaneous detection of GSTM1, GSTT1, and GSTP1 Ile105Val polymorphisms that can be used in most molecular biology laboratories.
Footnotes
Acknowledgments
The authors thank volunteers who donated blood samples for this study. This work was supported by the National Natural Science Foundation of China (no. 30860319, 81160105, 81160248) and the Project Foundation of Jiangxi Education Department (no. GJJ13165).
Author Disclosure Statement
No competing financial interests exist.