
Abstract
Aims: Point-of-care genetic analysis may require polymerase chain reaction (PCR) to be carried out on whole blood. However, human blood contains natural inhibitors of PCR such as hemoglobin, immunoglobulin G, lactoferrin, and proteases, as well as anticoagulant agents, including EDTA and heparin that can reduce whole blood PCR efficiency. Our purpose was to develop a highly specific, direct whole blood single-nucleotide polymorphism (SNP) analysis method based on allele-specific (AS) PCR that is mediated by Pfu DNA polymerase and phosphorothioate-modified AS primers. Results: At high Mg2+ concentrations, Pfu DNA polymerase efficiently amplified genomic DNA in a reaction solution containing up to 14% whole blood. Among the three anticoagulants tested, Pfu DNA polymerase showed the highest activity with sodium citrate. Meanwhile, Triton X-100 and betaine inhibited Pfu DNA polymerase activity in whole blood PCR, whereas trehalose had virtually no effect. These findings provided for the development of a low-cost, simple, and fast direct whole blood genotyping method that uses Pfu DNA polymerase combined with phosphorothioate AS primers for CYP2C9*3 and VKORC1(−1639) loci. Conclusions: With its high DNA amplification efficiency and tolerance of various blood conditions, Pfu DNA polymerase can be used in clinical laboratories to analyze SNPs in whole blood samples.
Introduction
P
Recent advances in medical genetics revealed an increasing number of disease-related single-nucleotide polymorphism (SNP) genetic variations, as well as SNPs that are associated with drug responses and severe adverse reactions (Hu et al., 2014). In this work we developed a highly specific direct whole blood SNP analysis method based on allele-specific (AS) PCR that is mediated by Pfu DNA polymerase and phosphorothioate-modified AS primers. The warfarin dosing-related CYP2C9*3 and VKORC1 (−1639) variants were used as proof of concept SNPs.
Materials and Methods
Enzymes and chemicals
Pfu DNA polymerase, uracil DNA glycosylase, dNTPs, and dUTP were purchased from Fermentas (Waltham, MA). PfuTurbo Cx was purchased from Thermo Scientific (Waltham, MA). Taq DNA polymerase was purchased from Sangon Biotech (Shanghai, China). Trehalose, Triton X-100, and betaine were purchased from Sigma (St. Louis, MO).
Primers
All primers were designed using the FastPCR 6 program (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). For each SNP, two forward primers matched to one of two alleles and a common reverse primer was designed. The synthesis of forward primers involved a phosphorothioate-modified nucleotide, wherein a nonbridging oxygen of the backbone phosphate was replaced by a sulfur atom at the 3′ end. Primer sequences and modification information are listed in Table 1. All primers were synthesized by Sangon Biotech.
Underlined letters represent unmatched bases. “s” indicates phosphorothioate modification. *1 allele (A) represents the CYP2C9 wild-type A gene; *3 allele (C) represents the CYP2C9 mutant C gene.
Blood sample collection and DNA extraction
Written informed consent was obtained from all patients in accordance with the Declaration of Helsinki and with an approval from the Ethics Committee of the First Affiliated Hospital, Soochow University. Genomic DNA was extracted from the blood samples using the UNIQ-10 Genomic Extraction Kit from Sangon Biotech. The DNA concentration was determined with a NanoDrop-1000 apparatus (Thermo Scientific, Shanghai, China).
Allele-specific PCR genotyping of genomic DNA using high-fidelity DNA polymerase and phosphorothioate-modified primers
For each SNP genotyping, two separate PCR reactions were performed. Each reaction contained an AS phosphorothioate-modified forward primer and a common reverse primer. The AS-PCR reaction was carried out in a 50 μL mixture containing 200 nM forward and reverse primers, 200 μM dNTP (dATP, dCTP, dGTP, and dTTP), 1X Pfu PCR buffer (Fermentas), 2.0 mM MgCl2, and 0.3 units Pfu DNA polymerase (Fermentas). Amplification was performed using a touchdown PCR program with the following steps: initial denaturation at 94°C for 5 min; 15 cycles of 94°C for 30 s, 68°C for 30 s with a 1°C decrease per cycle, and 68°C for 40 s; 25 cycles of 94°C for 30 s, 58°C for 30 s, and 68°C for 40 s; and final extension at 68°C for 2 min. PCR products were electrophoresed on a 2% agarose gel and visualized with 0.5 μg/mL ethidium bromide.
Direct whole blood DNA amplification with Pfu DNA polymerase
PCR reactions were carried out as described above. Instead of using genomic DNA, 0.5-20% whole blood was added directly to the reaction mixture. Various Mg2+ concentrations and Pfu amounts were used to optimize the amplification conditions. For the remainder of the experiments, 3.0 mM Mg2+ and 2.0 units Pfu were used for direct whole blood AS PCR. The PCR tubes were briefly centrifuged at 20,000 rpm, and 8.0 μL supernatant was loaded onto an agarose gel.
Direct whole blood PCR with uracil DNA glycosylase, dUTP, and PfuTurbo Cx
To eliminate PCR product contamination, the PCR conditions described above were modified by the addition of 1 unit UNG (Fermentas) and a dUTP mixture containing dNTP (200 μM of dATP, dGTP, dCTP, dTTP, and dUTP) to the 50 μL reaction volume. PfuTurbo Cx was used instead of Pfu. A 2 min incubation period at 50°C was added before the initial 5 min denaturing step of the touchdown PCR. The products from PCR reactions with or without UNG treatment were compared after electrophoresis on a 2% agarose gel.
Genotyping by direct DNA sequencing
Forty genomic DNA samples were amplified with sequencing primer sets specific for each SNP. PCR amplification was performed with Taq DNA polymerase at 94°C for 4 min; followed by 35 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s; and final incubation at 72°C for 4 min. PCR products were examined by electrophoresis on 2% agarose and sequenced by Huada (Shenzhen, China).
Results
CYP2C9 and VKORC1 genotyping by AS PCR with Pfu DNA polymerase and phosphorothioate-modified AS primers
To genotype warfarin dosing-related SNPs of CYP2C9 and VKORC1, we designed AS forward primers with the SNP site located at the 3′ end and tested them with several common reverse primers. Only the primer that produced a single PCR product was selected for further optimization. The genotype could be determined by generating a PCR product with the AS primer. Initially, only base −1 at the 3′ end was modified with a phosphorothioate bond. As in our previous study, Pfu DNA polymerase was used here as it had the best discrimination capability compared to Vent and Phusion DNA polymerases from New England Biolabs, and Pfu DNA polymerase from Fermentas. We first optimized the PCR conditions by modifying the DNA concentration, enzyme amount, and thermal cycling conditions. The best, although imperfect, allele discrimination was achieved using 40 ng DNA and 0.625 units Pfu DNA polymerase with a touchdown PCR cycling program in a 50 μL reaction volume (data not shown). Further optimization was performed by introducing a second phosphorothioate bond at base −2 at the 3′ end of all AS primers as well as a mismatched base in the two AS primers for VKORC1(−1639) (Table 1). With these optimized primers, perfect allele discrimination after the AS PCR was achieved (Fig. 1, Top). To confirm the accuracy of SNP recognition, the same genomic DNA samples were amplified with an outer 5′ primer and common reverse primers for CYP2C9*3 and VKORC1 (−1639). The resulting PCR products were purified and sequenced in both directions (Fig. 1, Bottom).

Pfu-mediated allele-specific (AS) polymerase chain reaction (PCR) genotyping of warfarin dosing-related single-nucleotide polymorphisms in CYP2C9 and VKORC1. Top panel, electrophoretic profile of all six warfarin dosing-related genotypes. Bottom panel, direct sequencing results. The genomic DNA of all genotypes was obtained from patient samples as reported previously [3], except for CYP2C9 *3/*3, which was obtained by direct DNA sequencing screening.
Effects of [Mg2+] on the efficiency of direct whole blood PCR by Pfu DNA polymerase
To test the effects of whole blood on PCR efficiency with Pfu DNA polymerase, we amplified the CYP2C9*1/*3 fragment directly from whole blood. For initial reactions we used the manufacturer's recommended buffer that contained 1.5 mM Mg2+ (Fermentas) with an increased amount of Pfu DNA polymerase relative to that used for genomic DNA. Effective amplification was observed for reaction mixtures with 1.0-4.0% whole blood (Fig. 2). We next tested a series of [Mg2+] that increased from 3.0 to 7.5 mM with different concentrations of whole blood that ranged from 0.5% to 18%. Higher [Mg2+] reversed the decline in amplification efficiency by blood (Fig. 2A). This anti-inhibitory effect increased concurrently with [Mg2+] to peak at 4.5 mM and decreasing above that concentration. At 4.5 mM Mg2+, effective amplification could be achieved in the presence of up to 14% whole blood. Interestingly, at each [Mg2+] concentration, strong amplification could be achieved with a broad range of blood concentrations (e.g., 4.0-12% at 4.5 mM Mg2+ and 2.0-8.0% at 3.0 mM Mg2+). Taking these results together with the finding that higher [Mg2+] decreases PCR specificity, we chose 3.0 mM Mg2+ for all subsequent experiments.
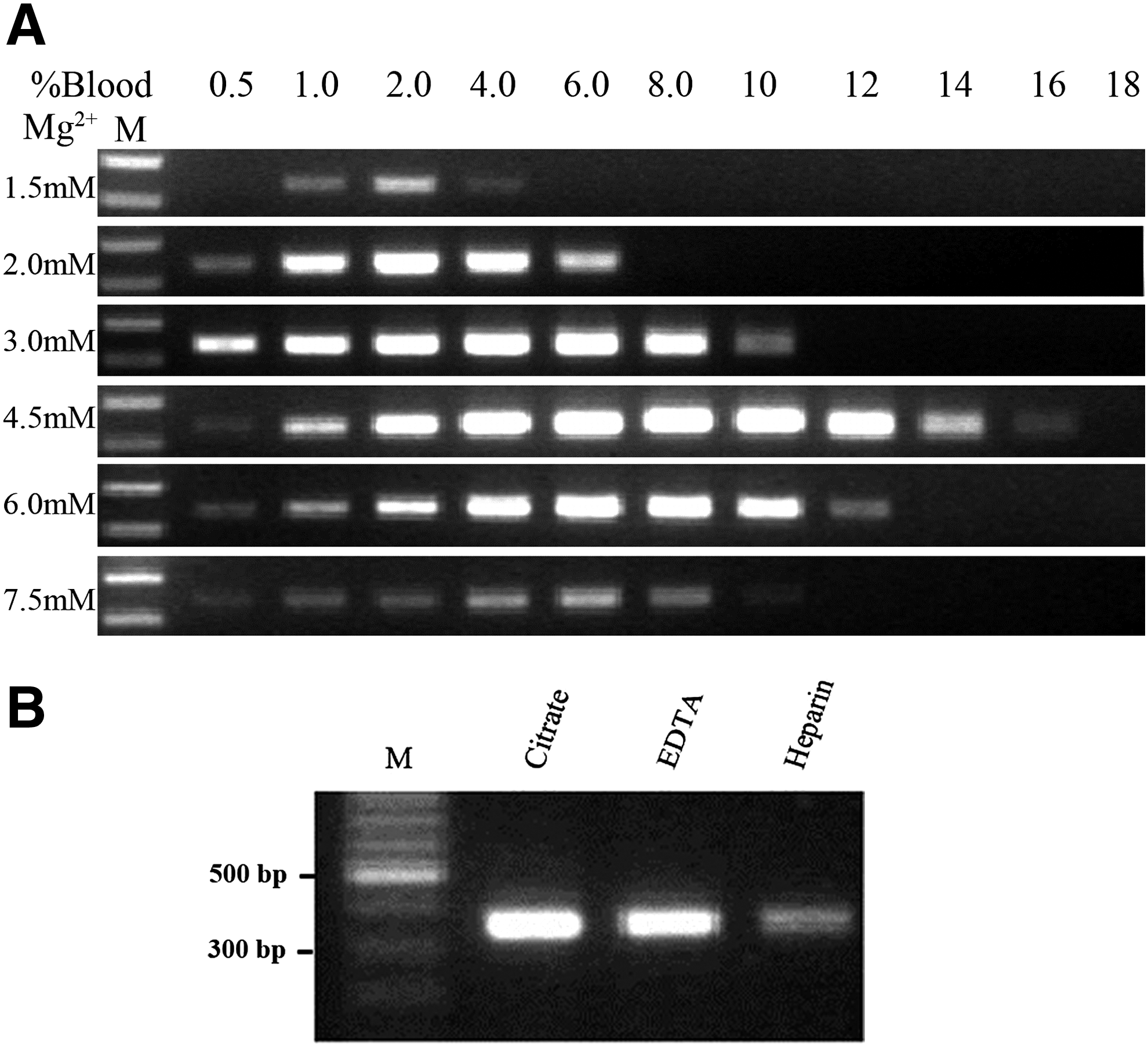
Effects of different anticoagulants on whole blood PCR amplification
Three different anticoagulants are commonly used in clinical blood samples: sodium citrate, EDTA, and sodium heparin. To compare the effects of these anticoagulants on direct Pfu DNA polymerase amplification, whole blood samples from a volunteer were treated individually with each of these three anticoagulants and the PCR efficiency using 3.0 mM Mg2+ and 4% blood was then compared. We found that PCR efficiency decreased in the following order from highest to lowest: sodium citrate > EDTA > heparin (Fig. 2B).
Effects of PCR additives on whole blood PCR mediated by Pfu DNA polymerase
Several chemicals are reported to enhance the efficiency of whole blood PCR with Taq DNA polymerase, including glycerol, trehalose, and betaine (Chakrabarti and Schutt, 2001; Jensen et al., 2010). We tested these chemicals in our Pfu DNA polymerase-mediated whole blood PCR system. Notably, the Fermentas Pfu PCR buffer contains 0.1% Triton X-100 at a working concentration. To test the hypothesis that high Triton X-100 concentrations can promote blood cell lysis, 0.5%, 1%, or 2% Triton X-100 was added to the PCR mixture for testing. Increasing Triton X-100 concentrations indeed significantly decreased the PCR product yield (Fig. 3A). Trehalose is reported to stabilize Taq polymerase, thus improving PCR efficiency (Fazekas et al., 2010). However, for Pfu DNA polymerase-mediated whole blood PCR, up to 10% trehalose only slightly enhanced the PCR yield (Fig. 3B). Meanwhile, betaine improves the PCR amplification efficiency of Taq polymerase by reducing the secondary structure of high GC (G: guanine; C: cytosine) content DNA fragments (Henke et al., 19997; Orpana et al., 2012). Betaine can also reverse the inhibitory effect of whole blood in Taq DNA polymerase-mediated PCR (Haqqi et al., 2002). However, for CYP2C9 and VKORC1 DNA templates, which have normal GC or high GC content, respectively, the addition of 1.0, 1.5, or 2.0 M betaine to Pfu DNA polymerase-mediated whole blood PCR did not enhance, but rather gradually decreased the PCR yield with increasing concentration (Fig. 3C).

Effects of PCR additives on the efficiency of direct whole blood PCR mediated by Pfu DNA polymerase.
Effects of whole blood on the uracil DNA glycosylase dUTP anticontamination system
In clinical genetic testing, uracil DNA glycosylase dUTP (UNG) must be used to treat the PCR mixture before thermal cycling. Since the Pfu-v93Q mutant of Pfu DNA polymerase is known to be resistant to dUTP inhibition, we tested whether UNG-dUTP can be used with direct whole blood PCR mediated by Pfu-V93Q DNA polymerase. We first synthesized an EGFP fragment (250 bp) that was amplified with dUTP instead of dTTP. Purified fragments (dU-DNA) with 3.7 × 108 to 3.7 × 1012 copies were used as templates in the presence of different concentrations of whole blood. The PCR reaction was incubated at 50°C for 2 min before the denaturation step. In the absence of whole blood, and with 3.7 × 1012 copies of dU-DNA input, only 10% of the product was seen following UNG treatment relative to untreated samples (Fig. 4, top). At template concentrations below 3.7 × 1012, UNG treatment completely degraded dU-DNA as no PCR product was obtained (Fig. 4, Top). There were no differences in the dU-DNA PCR reactions in the presence or absence of 4-12% whole blood (Fig. 4, middle panels). However, at 16% whole blood there was no PCR product with 3.7 × 1012 copies of input dU-DNA with UNG treatment and reduced amounts of PCR product without UNG treatment (Fig. 4, bottom panel). This result is likely because high concentrations of whole blood inhibited the activity of the mutant Pfu DNA polymerase. As shown in Figure 2, there was no PCR product for 12% whole blood with 3.0 mM Mg2+, which differs from that seen for samples that did not receive UNG treatment. This apparent discrepancy may be due to the excessive number of copies (∼107-108 copies of genomic DNA template) of exogenous DNA template used in this experiment as compared to the genomic DNA that was used as the template for the PCR reactions shown in Figure 2. Taken together, these results show that whole blood does not affect the activity of UNG.
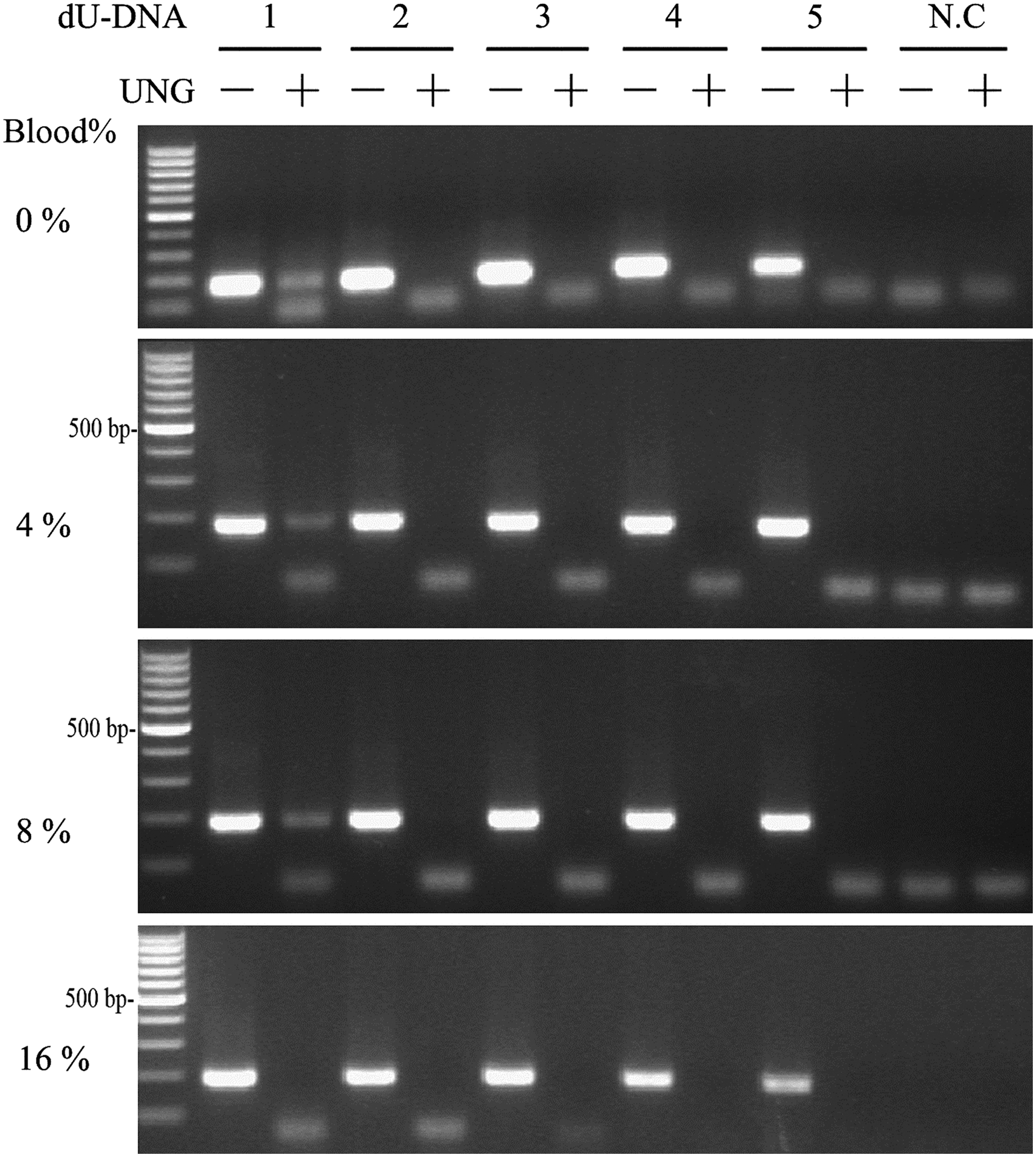
Effects of whole blood on the activity of uracil DNA glycosylase (UNG). The activity of UNG (1 unit) was tested in the presence of different concentrations of whole blood with sodium citrate as an anticoagulant and a synthetic nonhuman DNA fragment (EGFP) containing dUTP at different concentrations. dU-DNA: PCR-synthesized EGFP fragment containing dUTP. (1) 3.7 × 1012 copies; (2) 3.7 × 1011 copies; (3) 3.7 × 1010 copies; (4) 3.7 × 109 copies; (5) 3.7 × 108 copies; (N.C.) water control.
Effect of blood sample freshness and the number of white blood cells on whole blood PCR mediated by Pfu DNA polymerase
Fresh whole blood samples from three healthy volunteers that were treated with sodium citrate were tested alongside three stored whole blood samples. For both fresh and frozen blood samples, the experiment was carried out at 3.0 mM Mg2+ with the same scheme as shown in Figure 2. All three frozen blood samples were efficiently amplified from 1% to 10% whole blood concentrations (Fig. 2). However, for fresh whole blood samples, PCR products were observed on agarose gels only in the presence of 8% whole blood (data not shown; Fig. 5A, right panel). These results suggest that fresh whole blood is difficult to amplify. Thus, to reduce cell lysis and improve PCR efficiency, three additional heating-cooling cycles were added just before the initial long denaturing step. Among the several heating-cooling schemes tested, 95-50°C cycles greatly improved the PCR yield without sacrificing genotyping specificity (Fig. 5A, left panel). Other heating-cooling schemes, such as 95-60°C, improved PCR yields, but reduced genotyping specificity (data not shown).
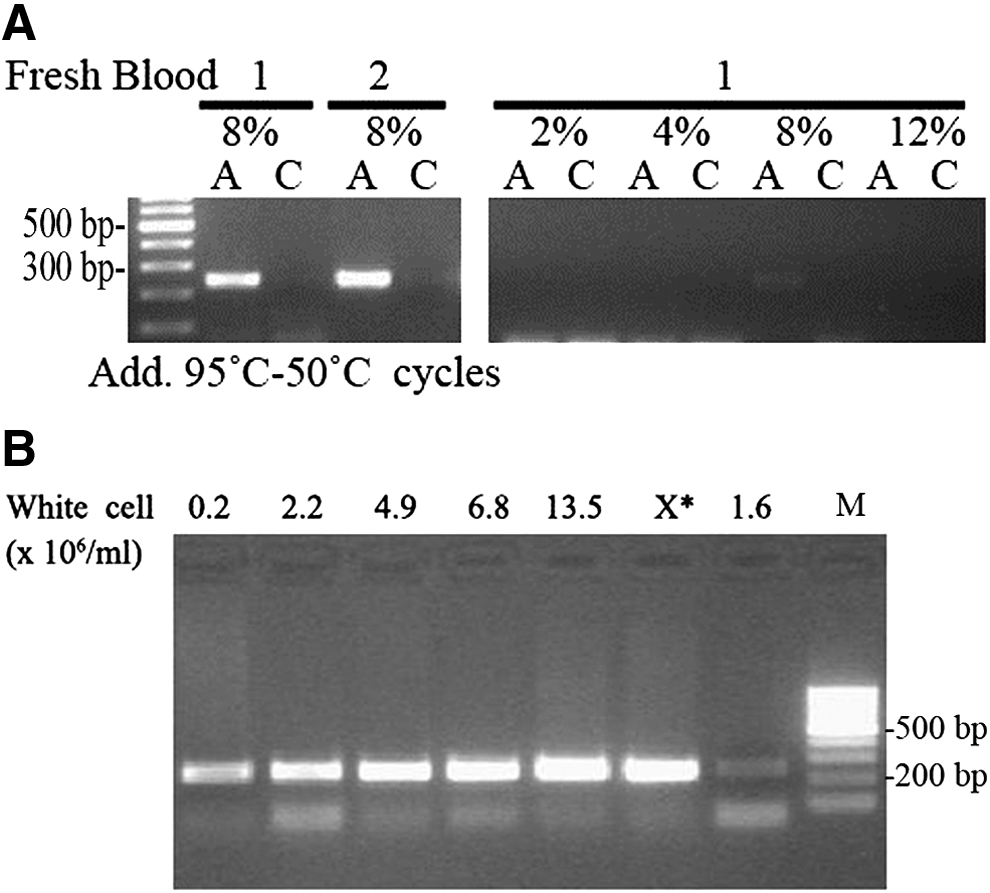
To test whether the white blood cell concentration affects the sensitivity of whole blood PCR mediated by Pfu DNA polymerase, PCR efficiency was compared among blood samples containing different number of white blood cells ranging from 0.2 × 106 to 13.5 × 106 cells/mL. For amplification of the CYP2C9*1/*3 fragment, we used the manufacturer's recommended buffer that contained 1.5 mM Mg2+ (Fermentas). In the presence of 4% whole blood, PCR product yields increased slightly with increasing white blood cell number (Fig. 5B). Even with a blood sample from a leukemia patient (labeled with “0.2”), which had high amounts of white blood cells, but only 800 copies of the DNA template, whole blood PCR still produced a fair amount of product (Fig. 5B). These results demonstrate that whole blood PCR has very high sensitivity and can tolerate wide variations in white blood cell numbers.
Direct whole blood genotyping of CYP2C9*1/*3 and VKORC1 (−1639) with Pfu DNA polymerase
To demonstrate that genotyping can be achieved by direct whole blood PCR with Pfu DNA polymerase, we performed genotyping screenings for CYP2C9*1/*3 and VKORC1 (−1639) with whole blood samples stored at −20°C. Whole blood AS PCR was carried out in the presence of 4% whole blood in a 25 μL volume containing 2 units PfuTurbo Cx, 0.5 units UNG, 3.0 mM Mg2+, 200 μM dNTP (dATP, dCTP, dGTP, dTTP, and dUTP), and 1X Fermentas PCR buffer. Touchdown PCR was performed with a 2 min incubation period at 50°C before the initial denaturation step. Genotyping analysis was performed after electrophoresis on a 2% agarose gel (data not shown). Fifty-nine blood samples treated with sodium citrate and EDTA could be genotyped for both CYP2C9 and VKORC1 (−1639), and 29 additional samples were genotyped only for VKORC1 (−1639) (Table 2). The whole blood genotyping results were confirmed by direct sequencing with 100% concordance.
“*1/*1” represents the Cyp2C9 wild-type homozygous AA genotype; “*1/*3” represents the Cyp2C9 mutant heterozygous AC genotype; “*3/*3” represents the CC genotype of the Cyp2C9 mutant homozygous gene.
Discussion
The concept of point-of-care genetic analysis in clinical genetic testing is now accepted by physicians as a means to provide simple, fast, and reliable genetic analysis of patient samples. Such analyses involve PCR amplification directly from unprocessed biological samples such as whole blood, but whole blood samples can be challenging to analyze as they contain substances that may inhibit the activity of DNA polymerases used for PCR amplification. In this study, we found that in the presence of high [Mg2+] concentrations, high-fidelity Pfu DNA polymerase can efficiently amplify genomic DNA in reaction mixtures that contain up to 14% whole blood. Moreover, Pfu DNA polymerase together with 3′ phosphorothioate-modified AS primers can mediate highly specific AS PCR for SNP genotyping (Zhang et al., 2003). Warfarin is the most commonly prescribed anticoagulant drug for the prophylaxis and treatment of venous and arterial thromboembolic disorders. CYP2C9 and VKORC1 have been identified as the underlying genes that are involved in warfarin metabolism and targeting (Yang et al., 2013). Warfarin dosing algorithms weighted according to genetic information for CYP2C9 and VKORC1 have recently been developed to provide more accurate information that can reduce the risk of warfarin overdose during the initial days of warfarin therapy (Krishna Kumar et al., 2013). CYP2C9*2, CYP2C9*3, and VKORC1 (−1639) are the major gene variants that are related to warfarin's effects. In the Chinese population, CYP2C9*2 is completely absent and the CYP2C9*3 and VKORC1 (−1639G) variants are rare. In our previous study, we genotyped CYP2C*3 and VKORC1 (−1639G) using the RFLP method and established a new warfarin dosing algorithm for Chinese patients. Although a number of warfarin genotyping assays are now FDA approved for use in clinical laboratories (Langley et al., 2009; Limdi et al., 2009; López-Parra et al., 2013), all of these assays require costly instruments and are time consuming.
Point-of-care genetic testing uses raw material such as whole blood for genetic testing that does not involve time-consuming laboratory processing for DNA extraction. Such testing is important for genetic analyses that are related to drug response, dosing, and severe adverse reactions since these tests greatly reduce the time needed to generate results, which in turn allows patients to receive prompt treatment.
In summary, we described a method for a direct whole blood genotyping method mediated by Pfu DNA polymerase and phosphorothioate-modified primers. As a proof of concept, this method was used to demonstrate that highly specific whole blood genotyping of warfarin dose-related SNPs, CYP2C9*3 and VKORC1 (−1639) could be performed.
Footnotes
Acknowledgments
This project was partially supported by the National Natural Science Foundation of China (No. 81271922), Natural Science Foundation of Jiangsu Province (BK20151194), Suzhou People's livehood Science and Technology (SS201511).
Author Disclosure Statement
No competing financial interests exist.