
Abstract
Introduction: Primary hyperoxaluria type 1 (PH1) is an autosomal recessive disorder caused by deficiency of alanine glyoxylate aminotransferase, due to a defect in the AGXT gene. Several mutations in this gene have been reported and some of them have been observed in multiple populations. The aim of our study was to analyze the mutations causing PH1 in the Moroccan population and to estimate its prevalence in Morocco. Methods: Molecular studies of 29 unrelated Moroccan patients with PH were performed by direct sequencing of all exons of the AGXT gene. In addition, to estimate the prevalence of PH1, we screened for the recurrent p.Ile244Thr mutation in 250 unrelated Moroccan newborns using real-time polymerase chain reaction. Results: Four pathogenic mutations were detected in 25 unrelated patients. The c.731T>C (p.Ile244Thr) was the most frequent mutation with a frequency of 84%. The other three mutations were c.33delC, c.976delG, and c.331C>T. The prevalence of the PH1 mutation among Moroccans was then estimated to range from 1/7267 to 1/6264. Conclusion: PH1 is one of the most prevalent genetic diseases in the Moroccan population and is probably underdiagnosed. Front line genetic testing for PH1 in Morocco should be initiated using an assay for the recurrent p.Ile244Thr mutation. This strategy would provide a useful tool for precocious diagnosis of presymptomatic individuals and to prevent their rapid progression to renal failure.
Introduction
P
PH1 (OMIM259900) is a rare autosomal recessive disorder due to deficiency of the alanine glyoxylate aminotransferase (AGT; EC2.6.1.44) (Danpure, 2001), a hepatic pyridoxine 5-phosphate-dependent enzyme, which catalyzes the conversion of glyoxylate to glycine. When AGT activity is absent, glyoxylate is converted to oxalate, which causes hyperoxalemia and hyperoxaluria with massive calcium oxalate deposition in the kidney and other organs (Coulter-Mackie et al., 1993). Individuals with PH1 present recurrent nephrolithiasis, nephrocalcinosis, and progressive deterioration of renal function with a history of renal stones or calcinosis. Age at onset of symptoms usually varies from 1 to 25 years. Approximately, 20% of affected individuals present severe early-onset disease (before the age of 6 months) often associated with growth delay, nephrocalcinosis, anemia, and metabolic acidosis. A half of patients present symptomatic nephrolithiasis in late childhood or early adolescence. Early death is common; 50% have end-stage renal disease at diagnosis (Coulter-Mackie et al., 1993; Coulter-Mackie and Rumsby, 2004; Cochat et al., 2011; Hoppe, 2012).
The PH1 phenotype is caused by mutations in the AGXT gene, located on chromosome 2 (2q37.3) and spanning over 10 kb DNA. AGT is a 392-amino-acid protein with a molecular weight of 43 kDa. More than 160 mutations throughout the 11 exons of the AGXT gene have been documented (www.hgmd.cf.ac.uk, accessed 2014) (Williams et al., 2009; Kanoun et al., 2013).
Diagnosis of PH1 is usually confirmed by measurement of direct AGT enzyme activity on tissue obtained on liver biopsy (Rumsby et al., 2004; Hoppe, 2012). Recently, advances in genetic testing have allowed new and noninvasive strategy for diagnosis. Polymerase chain reaction (PCR), based on the amplification of DNA extracted from peripheral blood lymphocytes, has been used to sequence the AGXT gene (Williams et al., 2009; Coulter-Mackie, 2005).
PH1 is determined by homozygosity or heterozygosity of two combined mutations in the AGXT gene. Two polymorphic variants characterize AGXT: the most common allele is called major and less frequent allele is the minor. The minor allele differs from the major by three polymorphisms: two substitutions, p.Pro11leu and p.Ile142Met, and a duplication of the allele 74Pb in intron 1. The minor allele is found in 20% of European and North American control population, but its frequency increases to 50% in patients of PH1. Only the p.Pro11Leu polymorphism has a phenotypic effect. It reduces the catalytic activity of AGT by a factor 3. The homozygosity redirects 5% of the protein in mitochondria and facilitates deleterious effects of mutations found in the common PH1 (Coulter-Mackie et al., 2003; Williams and Rumsby, 2007).
Genetic analysis of AGXT gene can detect mutations in most of suspected patients, supplanting the need for a liver biopsy. Four mutations, p.Phe152Ile, p.Gly170Arg, p.Ile244Thr, and p.Lys12Glnfs*156, represent more than 50% of PH1 alleles and represent the basis of genetic screening for PH1 (Monico et al., 2007). Some mutations are found in specific ethnic groups, like the p.Ile244Thr mutation with a North African/Spanish origin (Santana et al., 2003; Lorenzo et al., 2006; Williams et al., 2009). This work is the first study realized in Morocco to analyze the mutations causing PH1 and to estimate the prevalence of the p.Ile244Thr mutation in Moroccan patients with PH1.
Patients and Methods
Patients
Twenty-nine Moroccan subjects were enrolled in this study from 22 inbred families with a diagnosis of PH. The study was approved by the Local Ethics Committee (Institut National d'Hygiene, Rabat). Each patient was informed about the aim of the study, and the consent to genetic testing was obtained. The diagnosis of PH was based on clinical findings (urolithiasis, nephrocalcinosis, and end-stage renal failure), elevated plasma oxalate, and urinalysis (raised oxalate) in combination with spectrophotometric analysis of the calculation. The subtype of PH was not clinically identified. The average age of patients at referral was 11.66 years, ranging between 1 and 33 years.
Molecular approach
Genomic DNA was isolated from peripheral blood using the QIAamp DNA Blood Mini Kit (Qiagen, Inc.). All exons and flanking regions of the AGXT gene were amplified by PCR and screened for mutations by direct sequencing. The PCR conditions were carried out in a volume of 25 μL containing 100 ng of DNA, PCR (buffer 10×), 0.2 mM of each dNTP, 1.5 mM MgCl2, 0.5 μL of each primer, and 0.5 U Taq DNA polymerase (Invitrogen). All PCR primers and conditions are shown in (Table 1). PCR was performed in a thermocycler, which was arranged uniformly for all reactions as followed: initial denaturation step at 95°C for 5 min, denaturation step at 95°C for 30 s, annealing step differs according to the exon for 30 s, polymerization step at 72°C for 1 min, and final extension step at 72°C for 7 min. PCR products were then revealed on agarose gel through electrophoresis. Mutation screening was performed by direct sequencing using the BigDye Terminator Kit (Life Technologies) on an ABI prism 3130 DNA sequencer (Life Technologies). Obtained sequences were aligned to the AGXT reference genomic sequence (GenBank: NM_000030.2) using DNA variant analysis software (Mutation Surveyor® software).
Next, we screened for the p.Ile244Thr mutation in 250 Moroccan unrelated newborns' umbilical cords by real-time PCR (Applied Biosystems 7500 Fast Real-Time PCR Systems) using TaqMan probes (Applied Biosystems). Samples detected as heterozygous were confirmed by Sanger sequencing.
Results
Epidemiological and clinical data of PH1 patients
Our study was composed of 29 unrelated PH patients from different regions of Morocco: 9 female and 20 male patients, aged from 1 to 33 years. The majority of the patients (75%) were consanguineous (22 of 29). All were cousin-german. The age of onset ranges from 45 days to 26 years. Fifteen of 28 patients (53%) had a positive family history for recurrent urolithiasis. Fourteen of 28 patients had renal failure associated to nephrocalcinosis. The clinical data were not available for one patient (Table 2).
F, female; M, male; Min, minor; Maj, major; ND, not done; PH, primary hyperoxaluria.
Identification of mutations
Molecular diagnosis was performed first by direct sequencing of selected exons 7, 5, and 4 to investigate the common mutations: the Maghrebian mutation, p.Ile244Thr, and the Arab mutation, p.Gly190Arg and p.Gly170Ala (Nagara et al., 2013). In heterozygous and negative patients for tested mutations, we performed a molecular analysis of the remaining exons of AGXT gene.
Analysis of the coding regions and flanking intronic sequences of the AGXT gene shows the presence of four different mutations in 25 patients (Fig. 1). The p.Ile244Thr mutation is the most frequent in our cohort (21 homozygous and 3 heterozygous patients). Sequencing of exon 4 and 5 was normal. We found the heterozygous mutation in exon 2 (c.331C>T) in one patient. This nonsense mutation produces a change from the amino acid Arginine to stop codon (p.Arg111*). Two frameshift deletion mutations (c.976delG; p.Val326Tyrfs*15 and c.33delC; p.Lys12fs*34) have been detected in two other patients.
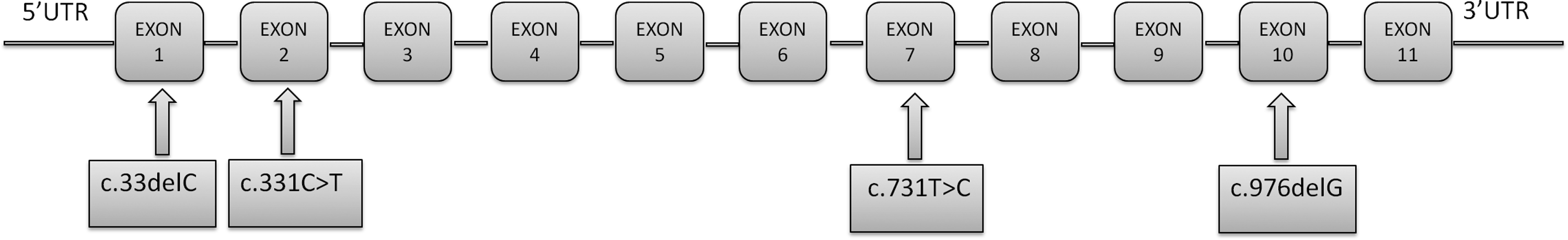
Schematic representation of the AGXT gene with the localization of the mutations found in this study.
The sequencing results showed the association among the following three intragenic polymorphisms, previously reported as the minor allele: the first one is a change in the c.32C>T allele, which encodes for leucine at codon 11 instead of proline. Other sequence changes on the minor allele were the presence of a 74-bp duplication in intron 1 in addition of IVS1+16A>G, c.166-56C>T, and c.1020A>G substitutions. We have also found another polymorphism in exon 10 (c.976G>A) (Table 3).
cDNA and protein numbering according to (ENST00000307503, NM_000030.2).
In four of our patients, no mutation was detected by Sanger sequencing of AGXT gene.
Prevalence of AGXT to the recurrent mutation p.Ile244Thr in Morocco
Frequency of heterozygous for the p.lle244Thr mutation was 4/250 (1.6%) and frequency of the mutated allele was 4/500 (0.8%).
The frequency of people who were heterozygous (2q) for this studied mutation was 1.6%, which enables us to estimate the prevalence (q2), under the assumption of Hardy-Weinberg, of patients homozygous or compound heterozygous for this mutation tested, and thus at risk to develop a PH1 to 1/15,625 (q2).
If we take into account the mean inbreeding coefficient of 0.0065 in the general population (Jaouad et al., 2009) and knowing that the prevalence of people who are homozygous according to the formula of Wright: P(MM): q2 + Fq(1 − q), with q the frequency of the mutated allele and F the mean inbreeding coefficient of the Moroccan population, based on the AGXT mutation prevalence which is 0.8/100, the expected frequency of biallelic AGXT mutations would be as follows:
The proportion of homozygous for the recurrent mutation p.lle244Thr among PH1 patients was 84%, considering that nonmutated patients have either PH1 with no mutation detected by Sanger sequencing (PH1 due to rearrangements of AGXT gene) or that they have another subtype of PH (UGlycolate analysis to distinguish PH1 and PH2 not available in Morocco).
Then, the estimated prevalence of hyperoxaluria type 1 considering the two hypotheses would range from 1/7267 to 1/6264.
Discussion
PH1 is a rare, recessive, inherited inborn error of glyoxylate metabolism. It remains underdiagnosed because of the large variability in its clinical presentation and age of onset. The estimated prevalence varies from 1 to 3 in 106 in Europe and North America (Leumann and Hoppe, 2001; Hoppe, 2012). The incidence of PH1 is low in these populations with a low level of consanguinity. It is ranging from 1.05/106 to 2.9/106 in France, Switzerland, and the Netherlands (Cochat et al., 1995; Kopp and Leumann, 1995; van Woerden et al., 2003). However, an increased frequency of PH1 has also been reported in Middle East countries because of the high rate of consanguinity in these populations (Rinat et al., 1999; Madani et al., 2001; Al-Eisa et al., 2004). It was estimated to be 5.5/106 in the Tunisian population (Belhaj et al., 2011; Nagara et al., 2013). The epidemiology of PH1 in Morocco is poorly documented compared to its neighbors (Tunisia and Algeria), probably because of misdiagnosis of the disease and lack of genetic studies. No data are available to date on PH1 prevalence in Morocco. Based on this study, the estimated prevalence of PH1 in Morocco is from 1/7267 to 1/6264. This prevalence classifies the PH1 among the most frequent diseases in Morocco. This rate is higher than other countries with a high rate of consanguinity as Tunisia. This result could be explained by the fact that the Tunisian study was performed using classic epidemiology disregarding the mutational spectrum of AGXT gene in Tunisian PH1, whereas in this study, the prevalence was calculated using epidemiological tools considering mutations of AGXT in Moroccan patients.
No study was conducted concerning the AGXT molecular pathology in the native Moroccan population. In our patients, the diagnosis of PH1 was initially based on clinical findings, elevated plasma oxalate, urine analysis (raised oxalate), and spectrophotometric analysis of the calculation urine showing a pure calcium oxalate monohydrate (whewellite). However, in our study, the subtype of PH was not identified clinically because analysis of UGlycolate and UGlycerate (defining the subtype of hyperoxaluria) is not available in Morocco.
In our cohort of 29 patients, we primarily screened exon 7 by direct sequencing. This strategy allowed us to identify genetic basis in 24 patients (42/50; 84%). Its frequency was considerably high such as frequencies reported in previous studies of other population backgrounds such as Tunisian, Spanish, and North African.
We have also identified two AGXT mutations: two single base deletions inducing frameshifts. The first one was c.33delC, found in the heterozygous state in one patient.
The second mutation was c.976delG, found in the heterozygous state in two unrelated patients (2/36, 5.55%). These mutations are a single-base deletion leading to a premature stop codon, predicting the consequent synthesis of a truncated protein. These deletions occurred in a compound heterozygous state with (c.731T>C) p.lle244Thr mutation.
The nonsense c.331C>T mutation was identified in one patient in the homozygous state. It causes the substitution of arginine by a premature stop codon. Prediction analysis showed that this nonsense mutation should lead to an abnormal protein (p.Arg111*). This mutation was associated with the “major” haplotype, defined by the most frequent alleles of specific AGXT polymorphisms. The identification of this mutation increases the number of genotypes implicated in PH1 in Moroccan population and confirms the molecular heterogeneity of the disease.
Six polymorphic variants were detected (Table 3). Four of these variants are intronic (intron1): IVS1+16A>G, IVS1+74 bp, c.166-56C>T, and c.166-47T>C. Two other variants, c.1020A>G (p.Ile340Met) and c.976G>A (p.Val326Ile), are in exon 10 (Nagara et al., 2013).
In addition, the missense variant c.32C>T changes the cyclic amino acid proline 11 to the aliphatic amino acid leucine. This polymorphism c.32C>T (p.Pro11Leu) affects a highly conserved N-terminal extended region of AGT protein, which is important in the process of dimerization and stabilization of the two protein subunits. This substitution, associated to the p.lle244Thr mutation, was predicted to be highly damaging to protein function by PolyPhen-2 prediction software with a score of 1. Moreover, these alterations are conserved across all analyzed species by the alignment of the AGT proteins in several species.
In four of studied patients, no mutation in AGXT gene was detected. This result does not exclude the diagnosis of PH and could be explained either by the fact that these patients have another subtype of PH or that they hold AGXT mutations undetectable by Sanger sequencing.
Concerning treatment, our patients systematically received conservative measures, including hydration, crystallization inhibitors, and pyridoxine. It was reported that in pyridoxine-sensitive patients, an improvement in renal function and a decrease in plasma and urine oxalate with high-dose vitamin B6 therapy were noted (Hoppe and Langman, 2003). Once the oxalate-clearance capacity of the kidneys is exceeded, calcium oxalate deposition becomes widespread and life-threatening, unless liver and kidney transplantation is performed. Early treatment of presymptomatic patients is possible and may prevent further loss of renal function.
Conclusion
PH1 is a clinically and genetically heterogeneous disorder. DNA sequencing of exon 7 essentially and then the exons 1 and 10 of the AGXT gene in this study can provide a useful, cost-effective, and first line investigation in Moroccan PH1 patients. This process may facilitate the molecular diagnosis of PH1 in our country and would allow an accurate tool for a precocious diagnosis in affected families (even without a clinical subtyping of PH) and detection of presymptomatic individuals. This will also prevent rapid progression to renal failure.
Footnotes
Acknowledgment
The authors thank the patients and their families for their collaboration.
Author's Contribution
B.L., M.T., and J.L. carried out the molecular genetic studies and drafted the manuscript. N.O., H.A., and K.S. provided clinical data. F.Z.L. helped in the real-time PCR. S.C.E. helped to draft the manuscript. A.S. participated in the design of the study and in the draft of the manuscript.
Author Disclosure Statement
No competing financial interests exist.